一個中國X連鎖腎上腺腦白質營養不良家系發生ABCD1基因突變(Ser342Ter)
2019-03-01 00:56:18胡琳晢南善姬隋冉冉卓明星
中國實驗診斷學 2019年2期
關鍵詞:基因突變
胡琳晢,南善姬,隋冉冉,卓明星,吳 杰
(吉林大學第二醫院 神經內科,吉林 長春130041)
X連鎖腎上腺腦白質營養不良(X-Linked adrenoleukodystrophy,X-ALD)是一種神經變性疾病,發病率大約為1∶15,000-30,000男性。該疾病的生化特征為飽和極長鏈脂肪酸(VLCFA)在腦白質、腎上腺和皮膚成纖維細胞中病理性地大量沉積。X-ALD的VLCFA的沉積與ABCD1基因突變有關[1]。我們現報道一個發生未發表的ABCD1基因突變(Ser342Ter)的中國X-ALD家系。
1 臨床資料
先證者1:男,49歲,因記憶力減退6個月,加重2個月于2014年2月28日入院。患者入院前6個月無明顯誘因逐漸出現記憶力下降,近2個月記憶力減退進行性加重,情緒容易激動,暴躁。既往外傷后右耳耳聾7年;吸煙史25年,30支/日,飲白酒6個月,飲酒量不詳。入院查體:血壓120/80 mmHg,神志清晰,言語流利,概測定向力、記憶力、計算力差,共濟失調,余神經系統查體未見陽性體征。血清皮質醇測定:皮質醇5點-7點125.50 ng/ml(66-268);促腎上腺皮質激素 55.10 pg/mL (0.10-46);皮質醇15點-17點 77.06 ng/ml(22-154)。醛固酮測定(臥位) 63.80 pg/mI(59.5-173.9),醛固酮測定(立位) 90.21 pg/mI(65.2-295.7),余血液常規生化檢查正常。心電圖、胸片檢查正常。頭部MR:腦干、雙側小腦半球、雙側側腦室后角旁可見斑片狀長T1長T2信號(圖1)。腎上腺CT:形態未見明顯異常。臨床診斷為腎上腺腦白質營養不良。住院期間,患者走路不穩、欣快、情緒容易激動,睡眠不良,便秘,4天后癥狀無明顯改善出院。患者出院后癥狀進一步加重,脾氣暴躁,開始酗酒。4月份,因精神癥狀,家人將其送至精神病院治療數日。6月份,患者到北京協和醫院檢查代謝6項正常,極長鏈脂肪酸:二十二烷酸37.18 mg/l(11.52-28.18),二十四烷酸47.23 mg/l(10.68-28.72),二十六烷酸1.23 mg/l(0-0.5),C24/C22 1.27(0-1.24),C26/C22 0.03(0-0.02),確診為腎上腺腦白質營養不良。10月份隨訪,患者癡呆、走路不穩,家人述曾走失1次,日常生活全部靠家人照顧。
先證者2,先證者1的弟弟,男,45歲,走路不穩、脾氣暴躁1年于2014年9月28日到門診就診。自述9年前開始逐漸出現走路略不穩,但不影響日常活動。智能正常,能勝任會計師工作。查體:右側跟膝脛試驗略欠穩準,雙下肢膝腱反射亢進。頭部MRI:腦干、雙側小腦半球、雙側側腦室后角旁可見斑片狀長T1長T2信號。
隨訪該家系,共有3代人,13名成員。先證者的父親健在,身體健康,母親因腦血管病過世,生前無類似癥狀。除了2位先征者,其他兄弟姐妹、子女均無異常(圖2)。
簽署知情同意書后,采集先證者及其他家庭成員的外周血5 ml,進行ABCD1基因突變檢測[2]。檢測結果:先證者1(II3)1和先征者2(II5)ABCD1 外顯子2,c.1025C>A,Ser342Ter,無義突變;ABCD1 外顯子2,c.1047C>A,Val349Val,同義突變;ABCD1 外顯子6,c.1548G>A,Leu516Leu,同義突變。其中,ABCD1基因外顯子2,c.1025C>A(Ser342Ter)改變無報道,該位點改變導致編碼氨基酸提前終止,提示為致病突變(如圖3)。ABCD1 基因外顯子2,c.1047C>A(Val349Val) 和外顯子6,c.1548G>A(Leu516Leu)序列改變均為多態性位點。II4、III3、III5ABCD1 基因外顯子2,c.1025C>A,Ser342Ter,雜合突變,為致病突變攜帶者。其他家庭成員ABCD1基因外顯子2未發現序列異常。
2 討論
我們對1個X-ALD家系進行ABCD1基因檢測,發現一個目前為止尚未報道的突變c.1025C>A,Ser342Ter。該突變存在于第2個外顯子,使第342個編碼絲氨酸的密碼子TCG改變為TAG(終止密碼子),導致ABCD1基因提前終止翻譯,形成異常ABCD1蛋白。自從X-ALD數據庫建立,已經報道了超過1300種突變(http://www.x-ald.nl)。現在我們又增加了一種新的突變。
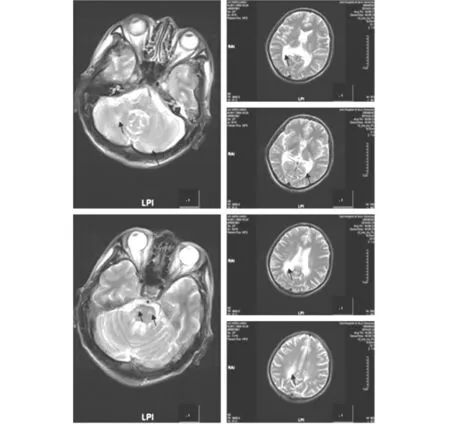
T2像示右側小腦、雙側腦干腹側,雙側側腦室旁點片狀高信號影
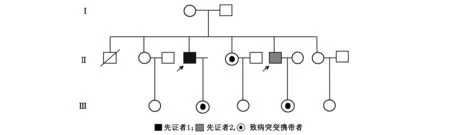
圖2 X連鎖腎上腺腦白質營養不良家系圖
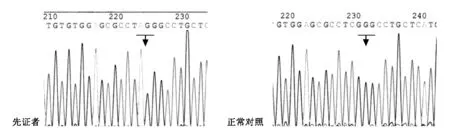
先證者1(II3)1和先征者2(II5)ABCD1 外顯子2,c.1025C>A,Ser342Ter,無義突變。↓ 示C突變為A
X-ALD有很多臨床表型,包括兒童腦型、青少年腦型、成人腦型、腎上腺脊髓神經病型(Adrenomyeloneuropathy,AMN)、Addison型、無癥狀型和雜合子型[2]。我們報道的這一家系中,先證者1發病半年后,病情迅速進展,符合腦型X-ALD(cerebral adrenoleukodystrophy,CALD)。 先證者2臨床表現符合AMN型,雖然頭部MRI顯示腦部脫髓鞘改變,尚未發生急性進展性的腦白質脫髓鞘。此外,
該家系中還存在3個攜帶者,目前為止尚無異常。
綜上,推測該家系的致病突變基因來自先證者的母親,她極有可能是該突變的攜帶者。女性攜帶者發病很少見,2位先證者的女兒均為攜帶者,建議進行相關方面的遺傳咨詢。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
