氣相色譜-質譜法同時測定高純三氯氫硅中4種甲基氯硅烷的方法研究
2019-02-20 05:38:10
分析儀器 2019年1期
(1.云南冶金云芯硅材股份有限公司,曲靖 655000;2.云南省光電子硅材料制備技術重點實驗室, 曲靖 655000)
三氯氫硅(沸點:33℃)是一種重要的硅化合物中間體,主要用于制造多晶硅或者有機硅材料。三氯氫硅中的主要含碳雜質是甲基氯硅烷[1],常見的4鐘甲基氯硅烷的化學性質極其活潑、沸點接近(一甲基二氯硅烷,沸點:41.9℃;三甲基一氯硅烷,沸點:57.7℃;一甲基三氯硅烷,沸點:66.4℃;二甲基二氯硅烷,沸點:70.5℃),在精餾工序中較難分離去除。因此,要控制多晶硅中碳含量,必須嚴格控制三氯氫硅中甲基氯硅烷的濃度。
三氯氫硅中4種甲基氯硅烷的分離具有相當的難度,一直是有機氯硅烷工業的難點和技術關鍵,至今沒有較好的檢測分析方法。俄羅斯標準采用傅里葉紅外光譜儀測定[2],其樣品處理操作條件非常嚴格復雜,容易對分析儀器及操作人員產生腐蝕和毒害,分析時間較長并且分析結果準確度差。國內一些多晶硅工廠采用氣相色譜法分析[3,4],但該方法檢測限不能滿足生產電子級多晶硅用高純三氯氫硅中碳含量的控制檢測要求。沈立俊[5]等采用氣相色譜-質譜(GC-MS)聯用技術,以1,2-二氯乙烷為內標,用內標法對三氯氫硅(SiH3Cl)中的含碳雜質甲基二氯硅烷快速分離并進行了分析測定方法研究。該方法僅對甲基二氯氫硅進行檢測,不能完全滿足電子級多晶硅生產過程對碳含量的控制需求。
本實驗采用氣相色譜-質譜(GC-MS)聯用儀,以1,2-二氯乙烷為內標,建立了三氯氫硅中雜質一甲基二氯硅烷(CH4SiCl2,略寫為MH)、三甲基一氯硅烷(C3H9SiCl,略寫為M3)、一甲基三氯硅烷(CH3SiCl3,略寫為M1)及二甲基二氯硅烷(C2H6SiCl2,略寫為M2)的快速分離與測定的方法,并對生產工藝中制得的三氯氫硅中的碳雜質進行的檢測。
1 實驗部分
1.1 儀器及試劑
1.1.1 儀器設備
氣相色譜-質譜聯用儀(Agilent 7890A/5975C GC-MS);石英毛細管柱:DB-1701型30 m×0.25 mm×0.25 μm;色譜進樣瓶:2 mL;微量注射器:1 μL,石英玻璃;容量瓶:100 mL,PFA材質。
1.2 試劑與材料
三氯氫硅,純度99.99%,密度1.35g/mL;四氯化硅,純度99.99%,密度1.50g/mL;辛烷,色譜純;一甲基二氯硅烷,純度99%,密度1.11 g/mL;二甲基二氯硅烷,純度99%,密度1.07 g/mL;一甲基三氯硅烷,純度99%,密度1.27 g/mL; 三甲基一氯硅烷,純度99%,密度0.86 g/mL;1,2-二氯乙烷,純度99%,密度1.25 g/mL;氦氣:純度不小于99.9995 %(φ);氮氣:純度不小于99.9995 %(φ)。
1.2 試樣準備
1.2.1 內標溶液的配制
內標貯存溶液A:1 mL溶液含1,2-二氯乙烷1 mg,取0.25 g的1,2-二氯乙烷置于250 mL的容量瓶中,用辛烷稀釋至刻度,搖勻。
內標溶液B: 1 mL溶液含1,2-二氯乙烷2 μg,移取1 mL內標貯存溶液,置于500 mL容量瓶中,用辛烷稀釋至刻度,搖勻。此溶液現用現配。
1.2.2 標準曲線的繪制
在通干燥氮氣的隔離操作箱中,分別準確移取一甲基二氯硅烷、三甲基氯硅烷、一甲基三氯硅烷、二甲基二氯硅烷0.1000g至100 mL容量瓶中,以高純四氯化硅稀釋至刻度,得到1000 μg/mL標準母液。然后根據需要分別移取10 mL、5 mL、1 mL、0.1 mL、10 μL的標準母液至100 mL容量瓶中,再分別加入50 mL的內標溶液,以高純四氯化硅稀釋刻度,分別得到100 μg/mL、50 μg/mL、10 μg/mL、1 μg/mL、0.1 μg/mL標準溶液,混勻。用微量注射器將1 μL試樣注入色譜儀,采集數據,繪制一甲基二氯硅烷、三甲基一氯硅烷、一甲基三氯硅烷、二甲基二氯硅烷標準曲線。
1.2.3 試樣溶液的制備
用1 mL注射器準確吸取0.5 mL內標溶液至色譜進樣瓶內,稱重,記錄質量為m0。將三氯氫硅試樣放入氮氣操作箱,吹干燥氮氣。移取試樣至該色譜進樣瓶并定容至1 mL,混勻,稱重,記錄質量為m1,色譜進樣瓶中三氯氫硅樣品質量為:
ms=m1-m0
1.2.4 測定
用微量注射器將1 μL制備好的試樣溶液注入氣相色譜-質譜聯用儀,記錄離子色譜圖。自工作曲線上查得的濃度相應的一甲基氫二氯硅烷、三甲基一氯硅烷、一甲基三氯硅烷、二甲基二氯硅烷的濃度(μg/mL)。
1.3 GC-MS分析條件
色譜條件:DB-1701石英毛細管柱(30 m×0.25 mm×0.25 μm);進樣口溫度為150℃;恒溫30℃,保持6min;載氣為高純氦氣(99.999%),恒定流速1.5mL/min;進樣量:1μL,分流進樣,分流比50∶1。內標法定量。
質譜條件:EI離子源,電離電壓70eV,輔助加熱溫度280℃,離子源溫度230℃,質量掃描范圍50~550u。采用選擇性離子檢測。溶劑延遲:三氯氫硅樣品,1.3 min。掃描離子見表1。
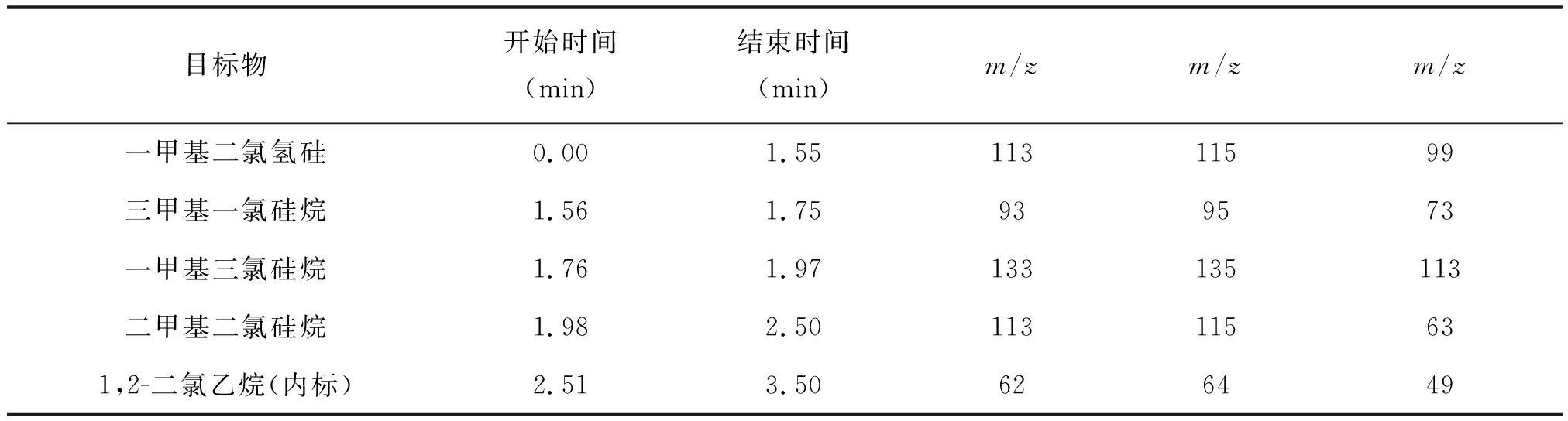
表1 掃描離子
1.4 甲基氯硅烷的定性與定量分析
采用標準樣品色譜圖和標準樣品加入的方法,通過對照保留時間進行定性,選取1,2-二氯乙烷為內標,采用內標法定量。
2 結果與討論
2.1 氣相色譜柱的選擇
由于4種甲基氯硅烷性質非常相近,沸點差別不大,分離有一定的難度。選用稍強極性的色譜柱,能有效增強組分與固定相之間的作用,有利于待測組分的分離,但由于甲基氯硅烷為弱極性物質,固定相極性不宜太強,因此本方法選用中等極性柱DB-1701,實驗結果證明,分離效果較好。
2.2 柱溫與載氣流速的選擇
分離的關鍵在于使組分能夠在柱中停留足夠長的時間,與固定相充分作用,因此需保持適當低的柱溫與載氣流速。經試驗選擇較低的初始溫度30℃,在保證GC-MS儀器正壓運行條件下選擇載氣流速1.5mL/min,使各組分得到了很好的分離。
2.3 分析方法的特性
2.3.1 保留時間與色譜圖
在已優化的儀器工作條件下,吸取濃度為1.0μg/mL的甲基氯硅烷混合標準溶液進行GC-MS分析,得到甲基氯硅烷混合標準樣品及內標物選擇離子色譜圖(圖1)。從圖1可以看出,4種甲基氯硅烷經毛細管柱分離在3min之內全部出峰,分析時間短并且各種甲基氯硅烷組分得到了很好的分離,本方法基線平穩,可很好地分析三氯氫硅/四氯化硅中的4種甲基氯硅烷。
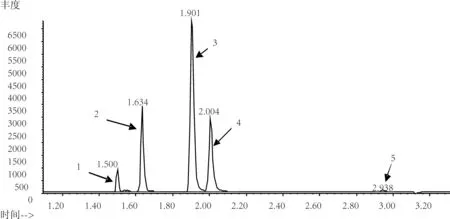
圖1 標準樣品甲基氯硅烷及內標物的選擇離子色譜圖1. 一甲基二氯硅烷;2.三甲基氯硅烷;3. 一甲基三氯硅烷;4. 二甲基二氯硅烷;5. 1,2-二氯乙烷。
2.3.2 標準曲線
將甲基氯硅烷標準混合溶液逐級稀釋,配制濃度為0.10、1、10、50、100μg/mL的標準工作液,按2.2中步驟進行樣品處理并按確定的儀器方法進行GC-MS分析,每個濃度標準溶液進樣3次,對各甲基氯硅烷相對于內標物的色譜峰面積比和濃度比進行回歸分析,校正曲線見圖2,相關系數見表2。
從表2可以看出,4種甲基氯硅烷工作曲線的相關系數都大于0.99,表明在所測定的濃度范圍內,待測物與其峰面積有很好的相關性。
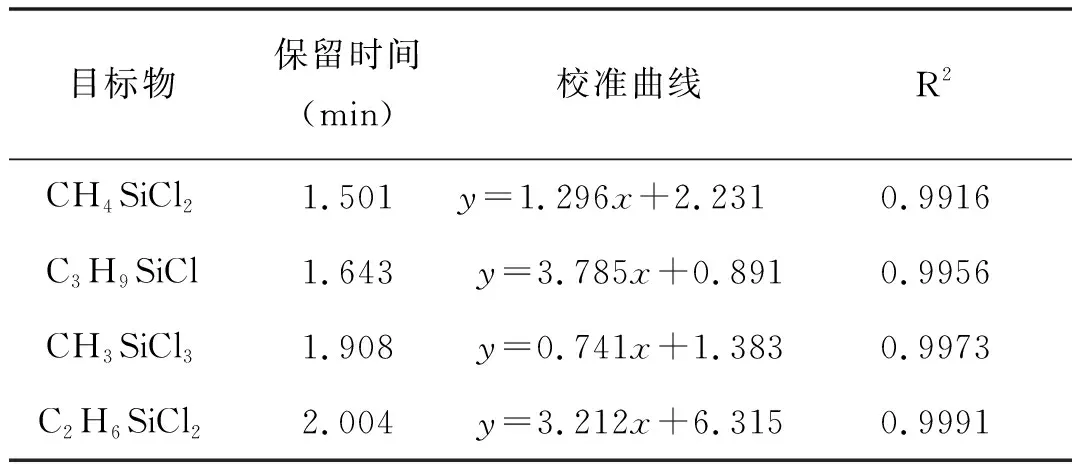
表2 甲基氯硅烷的校準曲線,相關系數以及檢出限
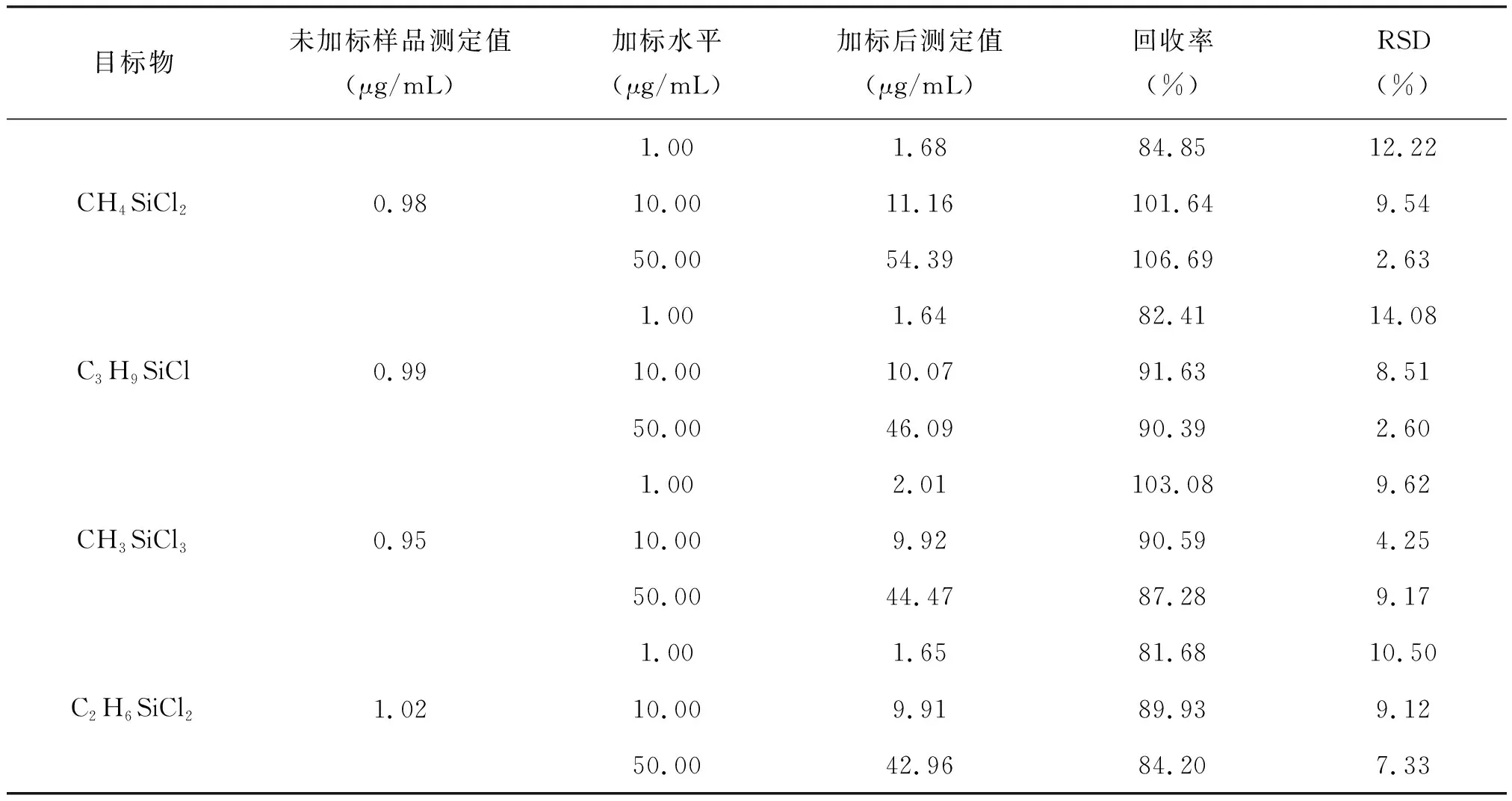
2.3.3 方法的回收率及精密度
取低、中、高濃度的甲基氯硅烷混合標準溶液分別加入三氯氫硅樣品中,然后與未加甲基氯硅烷標樣的三氯氫硅樣品,根據2.1節平行進行處理及色譜分析,各做6次求平均值,計算回收率,結果表明,4種甲基氯硅烷在四氯化硅種的回收率在81.68%~106.69%,相對標準偏差為2.60%~12.22%,見表3。該方法重現性好,可快速靈敏的測定三氯氫硅/四氯化硅樣品種4種甲基氯硅烷。
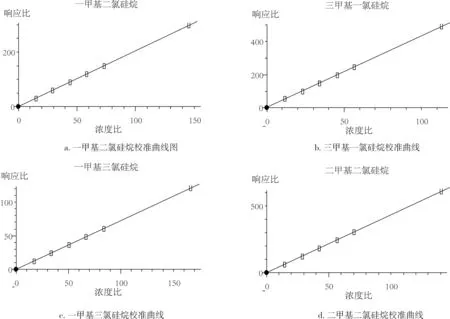
圖2 校準曲線
目標物未加標樣品測定值(μg/mL)加標水平(μg/mL)加標后測定值(μg/mL)回收率(%)RSD(%)CH4SiCl20.98 1.00 1.68 84.85 12.22 10.00 11.16 101.64 9.54 50.00 54.39 106.69 2.63C3H9SiCl0.99 1.00 1.64 82.41 14.08 10.00 10.07 91.63 8.5150.00 46.09 90.39 2.60 CH3SiCl30.95 1.00 2.01 103.08 9.6210.00 9.92 90.59 4.25 50.00 44.47 87.28 9.17 C2H6SiCl21.02 1.00 1.65 81.68 10.50 10.00 9.91 89.93 9.12 50.00 42.96 84.20 7.33
3 應用實例
生產線上取得的三氯氫硅樣品,按2.1節處理后進氣相色譜質譜分析,測定結果見表4。

表4 實際樣品測定結果
注:“-”低于儀器檢出限,未檢出。
4 結論
建立了氣相色譜質譜快速分離與測定多晶硅生產中原料三氯氫硅樣品及精餾三氯氫硅中甲基氯硅雜質的方法。本方法具有前處理過程簡單,分析時間短,重現性好,線性范圍寬等優點,適用于多晶硅生產工藝中三氯氫硅中的含碳雜質的檢測。
