吡格列酮對單鈉尿酸鹽誘導THP-1細胞炎癥反應的影響及其機制
2019-02-12 15:45:56楊珂珂楊興義陸靜徐玉梅
山東醫藥 2019年28期
關鍵詞:水平
楊珂珂,楊興義,陸靜,徐玉梅
(同濟大學附屬上海市第四人民醫院,上海200080)
目前已經明確痛風的病因為嘌呤代謝紊亂,即尿酸排泄減少或生成過多。當血液中尿酸持續升高,就會以單鈉尿酸鹽(MSU)的形式沉積于關節及臟器中,導致急性炎癥反應和病理損傷[1~3]。隨著生活水平的不斷提高,高尿酸血癥的發病率越來越高,特別是經濟發達地區和沿海城市,患者率高達23.5%[4]。但是治療痛風的手段非常有限,所涉及痛風急性發作期的藥物主要有三大類,即秋水仙堿、非甾體類消炎止痛藥物和糖皮質激素類,這些藥物長期應用,均存在一定的副作用。因此,發現新的干預靶點,對研發可適用于長期安全使用的新藥有著重大意義。過氧化物酶體增殖物激活受體γ(PPAR-γ)是由配體激活的核轉錄因子,是核素受體超家族中的成員, PPAR-γ活化可產生較強的抗炎作用[5]。吡格列酮是人工合成的高親和力PPAR-γ配體,因可改善胰島素抵抗,目前已廣泛應用于2型糖尿病的治療。已有研究表明吡格列酮作為PPAR-γ激動劑可使PPAR-γ活化從而減輕MSU誘導的大鼠急性痛風關節炎、大鼠急性腹膜炎和小鼠皮下氣腔炎癥[6,7],但具體機制尚不清楚。本研究采用體外實驗探討吡格列酮抑制炎癥反應的機制,以期為痛風提供新的治療靶點。
1 材料與方法
1.1 材料 人單核細胞(THP-1細胞)購自美國ATCC;DMSO培養基、佛波酯(PMA)購自美國 Sigma公司;FBS、EDTA購自美國Gibcog公司;吡格列酮(Piog,PPAR-γ激動劑)、T0070907(PPAR-γ抑制劑)、LY294002即蛋白激酶B(AKT)抑制劑購自美國Abmole公司;AKT抗體、p-AKT抗體購自美國Cell Signaling Technology;PPAR-γ抗體購自北京柯諾迪商貿有限公司;IL-1β ELISA試劑盒購自上海拓然生物科技公司;流式試劑盒購自美國Biolegend公司;MSU購自美國Sigma公司。6孔細胞培養板、T25培養瓶購自美國Corning公司;倒置顯微鏡、活細胞工作站購自萊卡纖維系統(上海)貿易有限公司;雙色激光ODYSSEY成像系統購自美國LI-COR公司;電泳槽和電轉儀購自美國Bio-Rad公司;酶標儀、實時熒光定量PCR儀、CO2細胞培養箱均購自美國Thermo公司。
1.2 THP-1細胞培養及分組 THP-1細胞于含10%FBS+1%P/S的RPMI1640培養基中培養,培養環境為37 ℃、5%CO2,每3~4 d換液1次。當細胞密度達到8×105/mL左右時,直接將細胞吸出,離心之后傳代。取大約1×107個細胞,用1 mL含有10%DMSO的FBS重懸,轉入凍存管,于-80 ℃冰箱過夜,第2天移入液氮中長期保存。將上述THP-1培養細胞分為PBS預處理組、T0070907預處理組、Piog預處理組,將上述三組細胞加PMA預處理24 h,分別以PBS(1%,500 μL)、T0070907(100 ng/mL,1 mL)、Piog(100 μg/mL,500 μL)預處理1 h,再各分兩組,一組不予干預,一組以MSU(100 μg/mL,1 mL)刺激16 h,繼續在37 ℃、5%CO2環境中培養,得到PBS組、T0070907組、Piog組、MSU組、MSU+T0070907組、MSU+Piog組。分別收集上清及細胞備用。
1.3 各組IL-1β標記陽性THP-1細胞構成比檢測 采用流式細胞術。取6支試管,每支試管加入上述各組THP-1細胞1×106個,然后加入50 μL染色緩沖液稀釋的IL-1β抗體2 μL,陰性對照管僅加入50 μL的緩沖液,暗處室溫孵育15~30 min。每管加2 mL 1×RBC Lysis Buffer 預熱至室溫,輕輕混勻。孵育10 min,離心,去上清,2 mL染色緩沖液洗1次,PBS洗1次,重懸細胞并渦旋振蕩。加入1 mL新鮮工作固定液,渦旋振蕩,孵育30~60 min,流式細胞染色緩沖液重懸細胞,上機分析,得到各組IL-1β標記陽性的THP-1細胞,計算IL-1β標記陽性的THP-1細胞構成比。
1.4 各組THP-1細胞上清液IL-1β水平檢測 采用ELISA法。按照試劑盒操作說明書進行操作,酶標儀于450 nm波長處測定每組的吸光度,建立標準曲線,根據標準曲線計算各組THP-1細胞上清液IL-1β水平。
1.5 各組THP-1細胞p-AKT、AKT、PPAR-γ蛋白表達檢測 采用Western blotting法。將體外培養的THP-1細胞分為四組,分別以PBS(1%,500 μL)、MSU(100 μg/mL)、Piog(100 μg,500 μL)、LY294002(100 μg/mL,500 μL)逐級干預,得到PBS組、PBS+MSU組,PBS+MSU+Piog組、PBS+MSU+Piog+LY294002組,37 ℃、5%CO2繼續培養16 h,棄去培養基,各組細胞加入RIPA裂解液裂解蛋白,BCA法測定蛋白濃度。每組取50~100 μg蛋白,SDS-PAGE分離蛋白,電轉至PVDF膜,用一抗和相應的熒光二抗孵育后,在ODYSSEY成像系統上進行曝光并記錄各組細胞中p-AKT、AKT、PPAR-γ蛋白相對表達量。
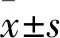
2 結果
2.1 各組IL-1β標記陽性THP-1細胞構成比比較 PBS組、T0070907組、Piog組、MSU組、MSU+T007090組、MSU+Piog組IL-1β標記陽性THP-1細胞構成比分別為(6.50±1.98)%、(10.76±1.39)%、(5.49±0.26)%、(72.56±2.73)%、(81.54±3.68)%、(10.80±2.26)%,與其他組比較,MSU組、MSU+T0070907組IL-1β標記陽性THP-1細胞構成比升高(P均<0.05)。PBS組、T0070907組、Piog組IL-1β標記陽性THP-1細胞構成比差異無統計學意義(P均>0.05)。
2.2 各組THP-1細胞上清液IL-1β水平比較 PBS組、T0070907組、Piog組、MSU組、MSU+ T007090組、MSU+Piog組THP-1細胞上清液IL-1β水平分別為(94.65±2.49)、(161.00±13.75)、(56.87±7.82)、(214.49±26.48)、(402.43±3.18)、(129.57±8.19)pg/mL。六組THP-1細胞上清液IL-1β水平差異有統計學意義(P<0.05)。MSU組、MSU+T0070907組IL-1β水平較PBS組、T0070907組、Piog組明顯升高,而MSU+Piog組較MSU組、MSU+T0070907組下降(P均<0.05)。
2.3 各組THP-1細胞p-AKT、AKT、PPAR-γ蛋白表達比較 PBS、PBS+MSU、PBS+MSU+Piog 、PBS+MSU+Piog+LY294002組p-AKT蛋白相對表達量分別為70.12±2.11、312.23±16.12、671.54±34.32、121.32±15.64;AKT蛋白相對表達量分別為123.14±8.92、243.32±15.86、435.15±43.56、116.26±7.86;PPAR-γ蛋白相對表達量分別為213.21±6.35、365.32±7.92、467.64±8.65、307.35±6.82;四組THP-1細胞p-AKT、AKT、PPAR-γ蛋白相對表達量比較差異有統計學意義(P均<0.05)。
3 討論
高尿酸血癥是痛風發病的基礎,隨著血尿酸升高,過飽和的MSU沉積在關節及組織,引起關節的急性炎癥反應,出現劇烈疼痛以及皮膚紅腫[8]。在急性痛風性關節炎發生發展過程中,各種炎癥細胞及細胞因子相互協同、誘生,伴隨炎癥過程始終[9]。沉積的MSU可趨化白細胞,白細胞吞噬MSU后迅速釋放白三烯和糖蛋白趨化因子,均成為痛風炎癥反應的重要介質。而單核巨噬細胞受MSU刺激后釋放白細胞介素,如IL-1β、IL-6、IL-8、腫瘤壞死因子α(TNF-α)等,均可加重痛風的炎癥反應。其中IL-1β是介導炎癥反應重要的炎癥因子[10]。IL-1β可激活巨噬細胞,增強其吞噬和殺傷作用。在滑膜、軟骨、關節滑液中均發現IL-1β的活性形式,被認為是炎癥調節的始動因素,亦是最經典的炎癥調節劑[11]。IL-1β還可刺激單核/巨噬細胞等合成IL-6[12]。本研究證實,MSU刺激THP-1細胞后,IL-1β水平明顯升高。
PPAR-γ在組織中分布很廣,最早發現在大腸、脂肪組織中表達[13]。隨后發現,PPAR-γ在血管平滑肌細胞、泡沫細胞、內皮細胞、單核巨噬細胞及腫瘤病灶中均異常表達[14]。在糖尿病模型大鼠中,吡格列酮可減少尿白蛋白的排泄,同時抑制巨噬細胞的浸潤和NF-κB活性等,進而減輕糖尿病腎病的組織病理學損害[15]。
本研究通過體外培養THP-1細胞,用MSU刺激細胞,并分別加入PPAR-γ激動劑吡格列酮及PPAR-γ抑制劑T0070907,證實吡格列酮可致炎癥因子IL-1β水平下降,而抑制PPAR-γ活性后,IL-1β水平又明顯升高,證實吡格列酮可減輕MSU誘導體外細胞的炎癥反應。然而吡格列酮發揮抗炎作用的具體機制目前尚不明確。
AKT是一種相對分子質量為57 kD的絲/蘇氨酸蛋白激酶,可被多種物質活化。AKT通過影響下游多種效應分子的活化狀態,發揮調節細胞存活、代謝、增殖作用。丁寶龍等[16]發現,吡格列酮可通過上調p-AKT的表達保護缺血再灌注的心肌細胞。本研究發現,吡格列酮干預后,PPAR-γ和p-AKT水平均明顯升高。體外研究亦證實,吡格列酮可上調THP-1細胞中PPAR-γ、AKT、p-AKT水平,當加入AKT抑制劑LY294002后,隨著AKT、p-AKT水平下降,PPAR-γ水平明顯下降,說明吡格列酮通過激活AKT途徑發揮抗炎作用。
綜上所述,PPAR-γ激動劑吡格列酮能有效抑制MSU誘導THP-1細胞的炎癥反應,其機制可能通過活化PPAR-γ/AKT通路實現。
猜你喜歡
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
火花(2019年12期)2019-12-26 01:00:28
人大建設(2019年6期)2019-10-08 08:55:48
人大建設(2019年12期)2019-05-21 02:55:32
雜文月刊(2018年21期)2019-01-05 05:55:28
人大建設(2017年6期)2017-09-26 11:50:44
學苑創造·A版(2015年11期)2016-01-14 09:03:27
俄羅斯問題研究(2012年1期)2012-03-25 09:54:45
中國火炬(2010年12期)2010-07-25 13:26:22
中國火炬(2010年8期)2010-07-25 11:34:30
