紫草素鈉脂質(zhì)體的制備及性能研究
2019-01-24 09:39:12施磊,葉雋,宋磊
藥學(xué)實(shí)踐雜志 2019年1期
施 磊,葉 雋,宋 磊
(1.上海市靜安區(qū)食品藥品檢驗(yàn)所,上海 200435; 2.上海市交通大學(xué)醫(yī)學(xué)院附屬同仁醫(yī)院,上海 200050)
紫草有消炎[1]、抗菌[2]、抗腫瘤[3]、抗病毒[4]等多種藥理作用,多用于治療褥瘡、尿布疹、皮膚燒傷、濕疹等各種皮膚炎癥,臨床應(yīng)用前景廣闊[5-6]。臨床研究發(fā)現(xiàn)紫草中的主要活性物質(zhì)紫草素(shikonin)不僅具有抗菌消炎和抗氧化等多種藥理作用,而且能夠促進(jìn)皮膚生長(zhǎng)和傷口愈合[7]。由于紫草素為脂溶性活性物質(zhì),因此目前常見的紫草制劑多為油性基質(zhì)的膏劑,活性成分易氧化變質(zhì),且吸收性和舒適度較差。此外,近年來(lái)研究發(fā)現(xiàn)紫草科植物中多含有吡咯里西啶類生物堿(pyrrolizidine alkaloids,PAs),這是一種肝毒性植物毒素,長(zhǎng)期或過(guò)量應(yīng)用會(huì)損害肝臟[8-10]。因此,歐美國(guó)家紫草相關(guān)制劑的應(yīng)用必須嚴(yán)格按照處方或醫(yī)囑。而我國(guó)目前市場(chǎng)上的大部分紫草相關(guān)制劑依舊直接以紫草提取物入藥,為臨床應(yīng)用帶來(lái)極大的安全隱患。
針對(duì)這些不足,近年來(lái)關(guān)于紫草素新型制劑的開發(fā)研究已有報(bào)道[11-12],但這些研究均是以紫草提取物為主藥,其成分復(fù)雜,含有較多雜質(zhì),不僅影響藥效,而且PAs的潛在危害依然存在。因此,基于這一現(xiàn)狀,本研究擬將紫草提取物提純,并將其以堿性有機(jī)鹽的形式制備成脂質(zhì)體,對(duì)其形貌、包封率和體外釋放等性質(zhì)進(jìn)行考察,為安全的新型紫草制劑的開發(fā)提供參考。
1 實(shí)驗(yàn)儀器及材料
1.1 儀器
沃特世E-2695液相色譜儀(沃特世科技有限公司),Hitachi H7650透射電子顯微鏡(HITACHI公司),PHS-3C型pH計(jì)(上海雷磁科學(xué)儀器股份有限公司),JY92-IIN超聲波細(xì)胞粉碎機(jī)(寧波新芝生物科技股份有限公司),ZS90納米粒徑電位分析儀(英國(guó)馬爾文公司)。
1.2 材料
左旋紫草素對(duì)照品(批號(hào),110769-200506,成都普思生物科技有限公司),紫草(上海市童涵春堂中藥店),甲醇(色譜純,德國(guó)Merck公司),其他試劑均為分析純。
2 方法與結(jié)果
2.1 紫草素的提取和提純
取5 g紫草粉末浸沒于60 ml 石油醚中, 65 ℃攪拌3 h,冷卻后過(guò)濾,加入新的石油醚60 ml重復(fù)提取2次。合并濾液,濃縮得紫草提取物粗品0.71 g,提取率為14.2%。將該粗品溶于60 ml石油醚中并加入等體積飽和碳酸鈉溶液,40 ℃條件下劇烈攪拌4 h后,分離上層有機(jī)液,加飽和碳酸鈉溶液萃取多次,直到下層不顯藍(lán)色,合并堿性水溶液,酸化后以乙酸乙酯萃取多次,直至上層無(wú)明顯紅色,合并有機(jī)層濃縮,得到純化的紫草素0.14 g提取率為2.8%(以紫草粉末質(zhì)量計(jì))。
以HPLC檢測(cè)紫草素提取物和提純后的紫草素,檢測(cè)結(jié)果見圖1:以左旋紫草素對(duì)照品為參照,由圖1C可知其出峰時(shí)間約為7.9 min,而紫草素提取物(圖1A)和堿化提純的紫草素(圖1B)也均在7.9 min左右出峰,可見二者主要成分為左旋紫草素。但將紫草素提取物與后者相對(duì)比,發(fā)現(xiàn)紫草提取物中含有更多雜峰,這是由多種雜質(zhì)引起的,而紫草素堿化提純后則非常純凈,其純度達(dá)到99.35%,除左旋紫草素之外,幾乎不含有其他成分。
2.2 脂質(zhì)體的制備
2.2.1紫草素脂質(zhì)體
精密稱取10 mg膽固醇,50 mg卵磷脂和5 mg紫草素粗提物溶于10 ml無(wú)水乙醇中超聲溶解,并將其用注射器勻速注入已水浴恒溫50 ℃ 的PBS溶液(pH 7.4,10 ml)中,反應(yīng)30 min后取出,將溶液置于旋轉(zhuǎn)蒸發(fā)器上35 ℃溫度下減壓蒸發(fā),除去有機(jī)溶劑。剩余溶液于50 ℃保溫1 h, 冷卻后超聲10 min,分別以0.45、0.22 μm的微孔濾膜過(guò)濾,得到堿化紫草素脂質(zhì)體,封裝并于4 ℃保存。用同樣方法制備空白脂質(zhì)體。

圖1 紫草提取物(A)、堿化紫草素(B)和對(duì)照品(C)的HPLC圖 A.紫草提取物;B.堿化紫草素;C.對(duì)照品;1.左旋紫草素
2.2.2紫草素鈉脂質(zhì)體
為提高藥物的體外釋放度,先將紫草素溶于少量飽和碳酸鈉中使之完全生成水溶性的紫草素鈉鹽,并以pH值為7.4的磷酸鹽緩沖溶液定容至10 ml。其他物料的加入以及制備方法與紫草素脂質(zhì)體制備方法完全相同。
2.3 脂質(zhì)體的外觀形態(tài)、粒徑和Zeta電位的測(cè)定
將脂質(zhì)體加適量超純水稀釋后制樣,在透射電子顯微鏡下觀察其外觀形態(tài),并用納米粒徑電位分析儀測(cè)其粒徑大小和Zeta電位,結(jié)果如圖2所示:粒徑分析結(jié)果顯示空白脂質(zhì)體的平均粒徑為98.11 nm,聚合物分散性指數(shù)(PDI)為0.117;而紫草素鈉脂質(zhì)體的平均粒徑為104.2 nm,PDI為0.103。兩種脂質(zhì)體粒徑大小均一,有明顯的雙分子層結(jié)構(gòu),均呈正態(tài)分布,且紫草素鈉脂質(zhì)體PDI值較小,說(shuō)明紫草素鈉脂質(zhì)體的分散度較好。由Zeta電位圖可以看出,空白脂質(zhì)體的Zeta電位為-15.0 mV,紫草素鈉脂質(zhì)體的Zeta電位為-14.8 mV,說(shuō)明以上兩種脂質(zhì)體的穩(wěn)定性相當(dāng)。將純化的紫草素以鈉鹽的形式制備成紫草素鈉脂質(zhì)體,與空白脂質(zhì)體相比粒徑有所增加,且中心不透明,這是由于紫草素堿化成鹽后親水性增強(qiáng),紫草素鈉脂質(zhì)體被包封在內(nèi)水相中引起的。

圖2 空白脂質(zhì)體(A、B)和紫草素鈉脂質(zhì)體(C、D)的透射電鏡圖和粒徑分布圖
2.4 左旋紫草素含量測(cè)定[13-14]
色譜條件:色譜柱:Inertisl?ODS-3(4.6 mm×250 mm,5 μm);流動(dòng)相:甲醇-0.025 mol/L磷酸(85∶15);流速:1.0 ml/min 檢測(cè)波長(zhǎng):516 nm;柱溫:25 ℃;進(jìn)樣量:20 μl。
標(biāo)準(zhǔn)曲線繪制:精密量取濃度為100 μg/ml的儲(chǔ)備液0.50、1.00、2.00、3.00、4.00、5.00 ml,分別于10 ml的容量瓶中用甲醇定容,配制成5、10、20、30、40、50 μg/ml的對(duì)照品溶液,高效液相色譜檢測(cè)并記錄峰面積A,繪制峰面積A-對(duì)照品質(zhì)量濃度C的標(biāo)準(zhǔn)曲線并進(jìn)行線性回歸分析,得到紫草標(biāo)準(zhǔn)曲線方程:A=3 429.474 2C-319.393 6(r=0.999 96)。結(jié)果表明紫草素在5~50 μg/ml范圍內(nèi)線性關(guān)系良好。
2.5 包封率測(cè)定
包封率是指包封在脂質(zhì)體內(nèi)的藥物量占投料量的百分比,是評(píng)定脂質(zhì)體制劑質(zhì)量的一個(gè)重要指標(biāo)。將脂質(zhì)體溶于pH7.4的磷酸鹽緩沖溶液中并定容至10 ml,量取3 ml加適量甲醇超聲破乳,再加甲醇定容至5 ml,并在12 000 r/min條件下離心10 min,取上清液,測(cè)其濃度為脂質(zhì)體中紫草素總量,記為C總(μg/ml);另取3 ml的紫草素鈉脂質(zhì)體混懸液在5 500 r/min條件下離心10 min,然后取上清液,測(cè)得未包入脂質(zhì)體的游離紫草素濃度C游(μg/ml),然后根據(jù)公式(1)計(jì)算其包封率。
包封率=(C總-C游)/C總100%
(1)
實(shí)驗(yàn)表明,紫草提取物脂質(zhì)體的包封率為20.4%(20.11%、20.42%、20.57%;RSD為1.2%),而紫草素鈉脂質(zhì)體的包封率為43.7%(44.02%、43.16%、43.84%;RSD為1.0%),與前者相比,紫草素堿化成鹽后包封率提高1倍。其原因主要是由于包封率測(cè)試是以左旋紫草素為參照,在藥物加入量相同的情況下,紫草素鈉脂質(zhì)體中紫草素純度更高,因此表現(xiàn)出較高的包封率。
2.6 體外釋藥特性
采用動(dòng)態(tài)透析法測(cè)定脂質(zhì)體的體外釋放特性:將脂質(zhì)體溶于pH7.4的磷酸鹽緩沖溶液中并定容至10 ml,并置于透析袋中,以含有0.5%吐溫-80的磷酸鹽緩沖溶液(pH7.4)為釋放介質(zhì),(37±0.5)℃下攪拌,分別于1/3、2/3、1、2、4、6、8、10、12、24 h取樣1 ml,同時(shí)補(bǔ)加相同溫度相同體積的釋放介質(zhì),測(cè)樣品中紫草素濃度,并按公式(2)計(jì)算藥物累計(jì)釋放量Q。
(2)
式中:Q為藥物累計(jì)釋放量,單位為%;Cn為第n次取樣的紫草素濃度,單位為μg/ml;Ve為取樣體積,單位為 ml;Md為脂質(zhì)體中包載的藥物質(zhì)量,單位為mg。
結(jié)果如圖3所示,12 h內(nèi)紫草提取物脂質(zhì)體及紫草素鈉脂質(zhì)體的藥物累計(jì)釋放量分別為48.0%(47.53%,48.12%,48.21%;RSD為0.8%)和65.8%(65.97%,66.12%,65.36%;RSD為0.6%),這是因?yàn)樽喜菟貕A化成鹽后水溶性更好,與脂溶性紫草素脂質(zhì)體相比,在磷酸鹽緩沖溶液中釋放更為徹底。

圖3 紫草素脂質(zhì)體(A)和紫草素鈉脂質(zhì)體(B)的藥物累計(jì)釋放曲線
3 討論
本研究首先對(duì)紫草素提取物進(jìn)行堿化提純。紫草素是紫紅色的蒽醌類脂溶性生物活性物質(zhì),因?yàn)榫哂蟹恿u基而顯酸性,其側(cè)鏈上的酯鍵在堿性條件下會(huì)水解,并與堿離子形成水溶性的紫草素鹽,其顏色為藍(lán)色,加入酸后,又可形成脂溶性的紫紅色紫草素,其轉(zhuǎn)化過(guò)程見圖4。
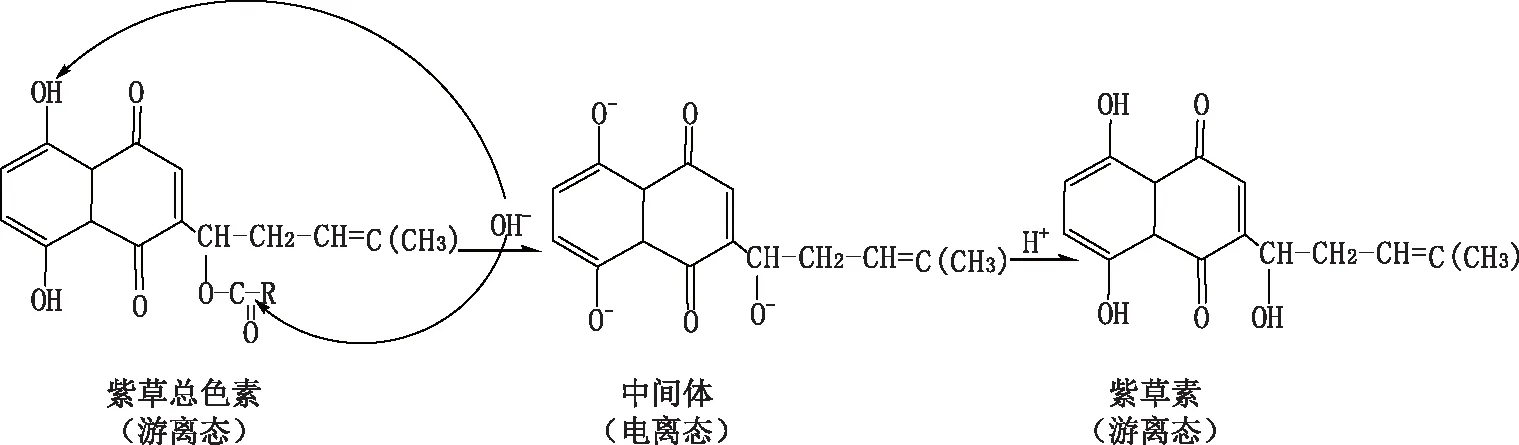
圖4 紫草素堿化過(guò)程
經(jīng)過(guò)堿溶酸沉法提純后,紫草素被有效純化,紫草提取物中大部分有害雜質(zhì)均被除去(圖1),其安全性大大提高,對(duì)解決現(xiàn)有市場(chǎng)上紫草相關(guān)制劑安全問題有積極意義。此外,對(duì)紫草素鈉脂質(zhì)體性質(zhì)研究發(fā)現(xiàn),與紫草素脂質(zhì)體相比,由于所含藥物純度和親水性更高,其脂質(zhì)體包封率和體外釋放度均比后者有較大提高,有利于提高藥效,為開發(fā)安全、高效的紫草新型制劑提供思路。
