基于密度泛函數理論的α-二亞胺鎳配合物催化乙烯/降冰片烯共聚合的計算化學研究
2019-01-21 01:24:48崔咪咪薛小松侯彥輝劉賓元
天津工業大學學報 2018年6期
楊 敏,姜 湃,崔咪咪,薛小松,侯彥輝,劉賓元
(1.河北工業大學 化工學院,天津 300130;2.南開大學元素有機重點實驗室,天津 300071;3.天津工業大學 材料科學與工程學院,天津 300387)
乙烯/降冰片烯共聚物被認為是最有前途的新型工業熱塑性聚合物,具有高玻璃化溫度、耐熱性、良好的光學透明性、低介電常數以及優異的防潮性能等,因此,其可以廣泛地應用于醫用材料、電子元器件和光學材料[1-3].由于乙烯/降冰片烯(E-N)共聚物的優異性能,使用不同種類催化劑催化乙烯/降冰片烯共聚反應的研究已經引起了越來越多的關注.乙烯/降冰片烯加成共聚物可以通過各類催化劑進行催化合成,如茂金屬催化劑[4-6]、前過渡非茂金屬催化劑[7-9]以及后過渡非茂金屬催化劑[10-12].另一方面,許多研究學者開始使用密度泛函理論(DFT)來研究乙烯/降冰片烯共聚反應[12-19]的反應機理.王永霞等[16]運用理論計算的方法發現在由半茂鈦催化的乙、降共聚過程中,降冰片烯單體的引入將降低初始插入步驟的反應能壘,促進共聚反應的發生.理論計算也被應用于探究金屬釩作為活性中心催化乙烯均聚、乙烯/降冰片烯共聚的反應機理[17].Tang等[18]運用密度泛函理論探究半茂鈦金屬配合物催化的乙烯/降冰片烯共聚反應機理,取得顯著成果.Kim等[19]運用密度泛函理論探究乙烯和降冰片烯在插入金屬活性中心過程中的競爭關系,發現降冰片烯單體上的環戊基展現出固定氫鍵的作用,加強降冰片烯單體插入活性中心的能力.
α-二亞胺鎳催化劑體系被廣泛地應用于乙烯均聚[20-24]、降冰片烯均聚[25-28]以及乙烯、降冰片烯嵌段共聚[29]反應中.然而,使用該催化體系催化乙烯和降冰片烯無規共聚反應以及運用密度泛函理論來探究該無規共聚過程的反應機理的研究未見報道.本課題組將N,N′-二(2,6-二異丙基)苊二亞胺溴化鎳催化劑應用于乙烯、降冰片烯均聚以及無規共聚反應,但研究結果發現該催化劑僅能催化均聚反應,而無法催化乙烯/降冰片烯無規共聚反應.為了探尋造成這一實驗結果的原因,本文使用密度泛函理論(DFT)方法模擬共聚過程中可能出現的所有反應結構,計算相應結構的勢能,并對乙烯和降冰片烯的引發反應、再插入反應以及乙烯的線性鏈增長和β-氫轉移反應分別進行能量對比.
1 實驗部分
1.1 實驗材料
所有對水、氧敏感的操作均在氬氣氣氛下的手套箱抑或是Schlenk線性管路中進行.N,N′-二(2,6-二異丙基)苊二亞胺溴化鎳參照文獻[30-31]進行合成,其平面結構如圖1所示;活化劑一氯二乙基鋁(AlEt2Cl,1.0 mol/L)、甲基鋁氧烷(MAO,1.5 mol/L),Albemarle公司產品;乙烯氣體,天津賽美特特種氣體有限公司產品;甲苯在使用前需經過鈉/二苯甲酮蒸餾干燥操作;氯苯在使用前經過氫化鈣干燥處理.所有其他的化學品均直接購買.
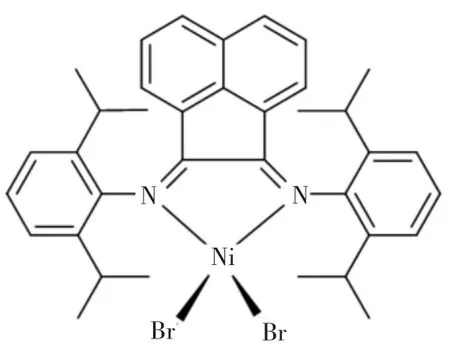
圖1 N,N′-二(2,6-二異丙基)苊二亞胺溴化鎳配合物的平面結構Fig.1 Planar structure of bis[N,N′-(2,6-diisopropylphenyl)imino] acenaphthene dibromonickel
1.2 乙烯、降冰片烯的均聚和無規共聚反應
聚合反應在100 mL具有機械攪拌功能的不銹鋼高壓釜中進行.在進行聚合實驗之前,高壓釜需在150℃下抽真空2 h,然后在氬氣環境下冷卻至室溫,最后用干燥的氬氣置換2次,乙烯置換1次.實驗時,在指定溫度下,依次注入溶劑甲苯、定量催化劑和助催化劑,充分混合后充入乙烯氣體達到指定壓力.反應結束時,使用10%酸化乙醇終止聚合反應.聚合產物使用蒸餾水和無水乙醇反復清洗3遍,置于60℃下的真空烘箱干燥24 h.
1.3 計算方法
對于聚合反應,反應過程的勢能和結構計算充分參照 Brookhart-Green[30]和 Cossée-Arlman[32]機理.本文所有的反應物、中間體、過渡態、產物的結構優化和能量計算均使用密度泛函數理論方法.反應過程中的所有吉布斯自由能的數值采用(SMD)B3LYP/[6-31G(d)+Lanl2dz]//(SMD)M06/[6-311++G(d,p)+SDD]水平進行計算.結構優化、振動頻率以及熱力學校正使用B3LYP方法,并對金屬鎳使用Lanl2dz贗勢基組,而其余所有原子使用6-31G(d)基組[33-34].甲苯溶劑的影響采用SMD溶劑化模型進行計算[35].所有結構優化的結構均采用頻率計算的方法進行過渡態抑或是穩定結構的驗證.
為了獲得更加精確的計算值,在(SMD)M06/[6-311++G(d,p)+SDD]水平下對已經經過(SMD)B3LYP/[6-31G(d)+Lanl2dz]水平優化過的穩定結構進行單點能計算.單點能計算中使用M06方法,對金屬鎳采用SDD贗勢基組,對其余所有原子采用6-311++G(d,p)基組.所有結構使用CYL軟件進行觀測.所有計算均使用Guassian09軟件進行,所有計算能量單位均使用kcal/mol(1 kcal=4.186 kJ).
2 結果與討論
2.1 α-二亞胺鎳配合物的催化活性
N,N′-二(2,6-二異丙基)苊二亞胺溴化鎳作為主催化劑A(catalyst A),甲基鋁氧烷或是一氯二乙基鋁做助催化劑,催化乙烯均聚、降冰片烯均聚和乙烯/降冰片烯無規共聚反應,研究不同催化條件包括反應溫度、反應時間、反應壓力以及鋁鎳比對催化劑活性所產生的影響,結果如表1所示.
由表1可以看出,在乙烯均聚反應中催化劑活性達到2.91×106g/(mol·h),在降冰片烯均聚反應中催化活性可達0.45×105g/(mol·h).然而在各種反應條件下,乙烯和降冰片烯無規共聚反應均無聚合產物.因此,取共聚反應后的反應液進行氣質聯用分析,譜圖顯示反應液中的主要成分是反應溶劑和反應單體.這個結果意味著該催化體系催化乙烯/降冰片烯無規共聚反應無法產生共聚產物.那么,為什么該催化體系能催化乙烯和降冰片烯均聚反應,而無法催化兩者的無規共聚反應?為了探究這些原因,本文采用密度泛函數理論計算方法,模擬共聚過程所有可能發生的反應結構并計算相應結構的能量.
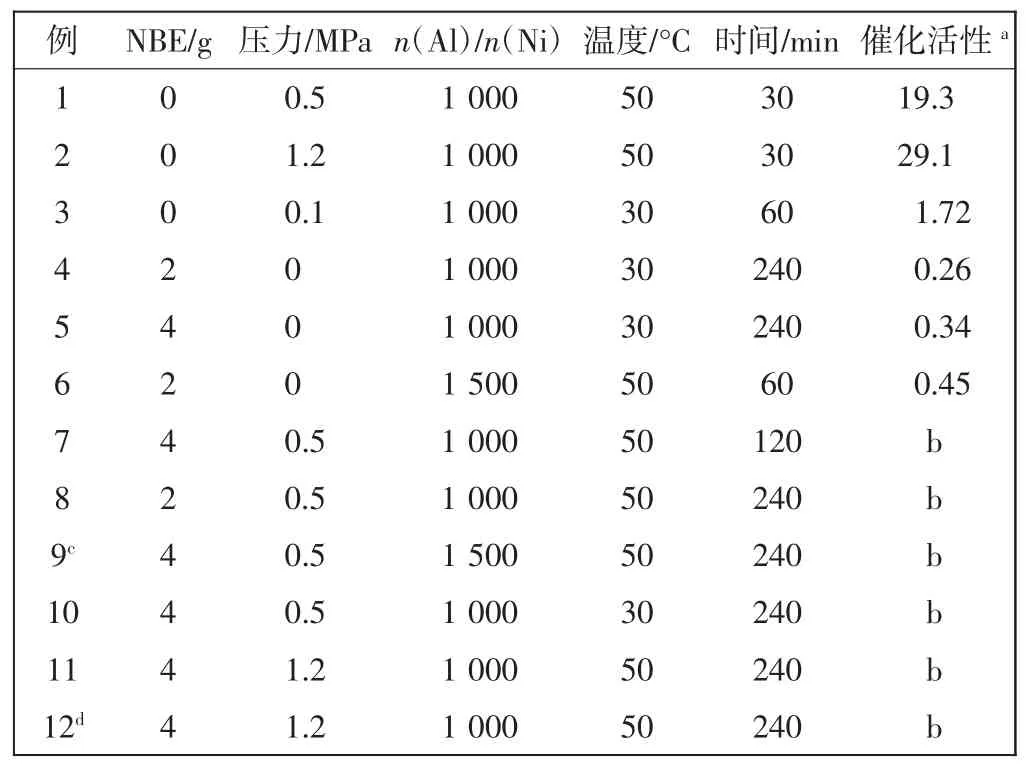
表1 催化劑A催化乙烯、降冰片烯均聚和共聚Tab.1 Homo-and copolymerization of norbornene and ethylene catalyzed with catalyst A
2.2 乙烯和降冰片烯插入Ni-CH3鍵過程對比
基于實驗結果,本文運用DFT理論研究降冰片烯和乙烯單體分別插入陽離子金屬活性中心的過程,模擬插入過程可能產生的結構并計算其對應結構的勢能,如圖2—圖4所示.
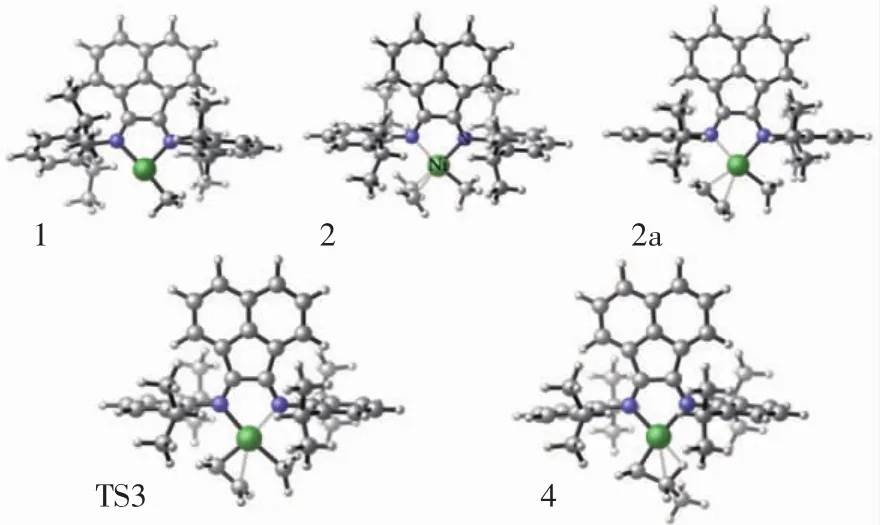
圖2 對于乙烯引發,陽離子引發結構1、配合物結構2/2a、過渡態結構3、產物結構4的分子結構Fig.2 For ethylene initiation,top view of molecular geometries of cation complex 1,π-complex 2,π-complex 2a,transition state 3 and product 4
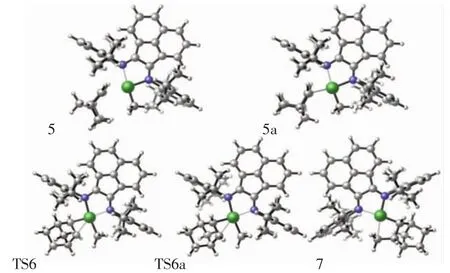
圖3 對于降冰片烯引發,配合物結構5/5a、過渡態結構6/6a、產物結構7的分子結構Fig.3 For norbornene initiation,top view of molecular geometries of π-complex 5,π-complex 5a,transition state 6,transition state 6a and product 7
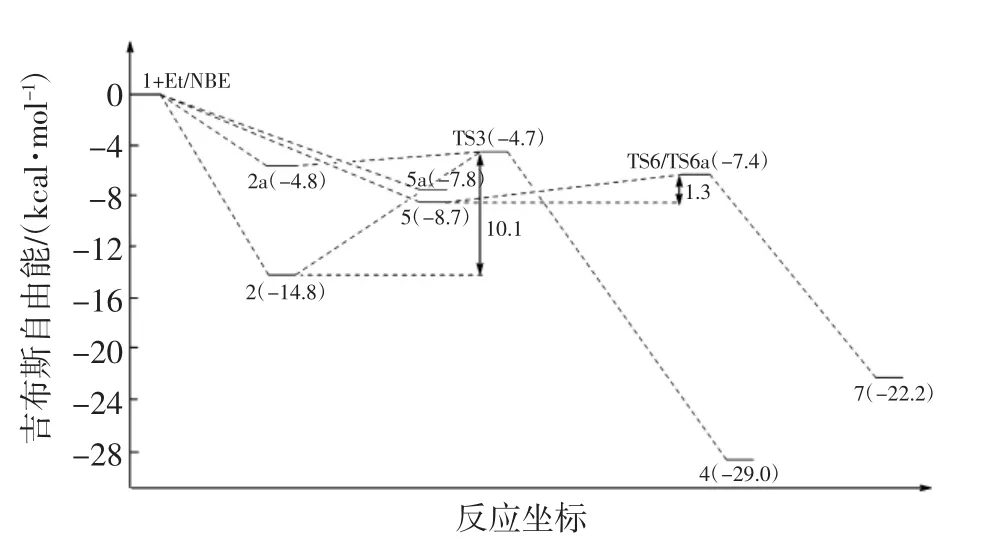
圖4 乙烯和降冰片烯插入陽離子活性中心勢能圖Fig.4 Calculated potential energy diagram for norbornene and ethylene insertion into Ni-CH3bond of[Ni(CH3)]+
根據文獻[30,36]的研究結果,可假設初始引發反應從陽離子復合物結構1開始,復合物結構2具有T型平面(Cs)結構且處于能量最低態,如圖2所示.乙烯插入陽離子活性中心的第一步是乙烯單體與復合物1進行配位.在配位過程將產生2種具有Cs對稱的π配位結構(2和2a),2種結構區別在于是平行或垂直于Ni(N=C—C=N)平面.從圖4所示勢能圖可以發現,復合物2的勢能比結構2a低10 kcal/mol,因此復合物結構2是更加穩定的結構.在反應過程中,結構2比結構2a更容易形成.形成配合物結構2后,經過渡態結構3(TS3)生成反應物結構4.Hessian分析證實結構3具有269.27i的虛頻,是乙烯插入陽離子活性中心過程的過渡態結構.基于圖4中的勢能基線,乙烯單體插入Ni—CH3鍵過程的總能壘為-4.7 kcal/mol.
降冰片烯插入陽離子活性中心的第一步是降冰片烯單體與復合物1配位形成配合物結構5,然后通過過渡結構6生成產物結構7,如圖3所示.對于降冰片烯單體與陽離子催化活性種的配位過程存在endo和exo 2種構型,因此,在降冰片烯插入陽離子活性中心的過程中也存在2種可能的反應方式.通過單點能計算,配合物中間體結構5a的能量為-7.8 kcal/mol,處于勢能淺井,結構穩定性遠不如勢能更低的中間體結構5,如圖4所示.因此,中間體結構5是更穩定的結構,并將優于結構5a率先形成.對于過渡態結構6(TS6)和 6a(TS6a),它們分別具有 237.92i和 238.16i的虛頻,但它們具有相同的勢能(-7.4 kcal/mol).綜上所述,降冰片烯插入陽離子活性中心反應的總能壘為-7.4 kcal/mol.
對比降冰片烯單體和乙烯單體插入陽離子活性中心反應的總能壘,發現降冰片烯單體在插入Ni-CH3鍵反應過程中具有更低的反應能壘,將更容易發生.因此,從理論上說明共聚過程中降冰片烯單體更占優勢,將比乙烯單體優先進行插入反應,此時體系中主要存在結構7.
2.3 乙烯和降冰片烯插入Ni—NBE結構過程比較
根據上述計算結果,將結構7作為再插入反應過程的初始結構,模擬再插入過程可能產生的結構并計算其對應結構的勢能,結果如圖5—圖7所示.
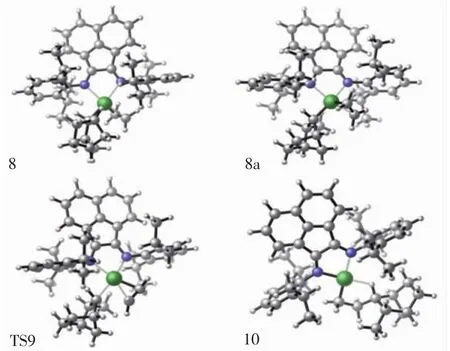
圖5 配合物結構8/8a、過渡態結構9,產物結構10的分子結構Fig.5 Top view of molecular geometries of π-complex 8,π-complex 8a,transition state 9 and product 10
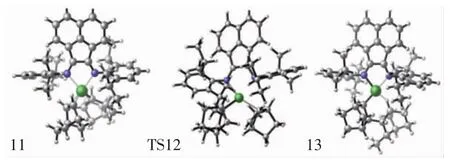
圖6 配合物結構11、過渡態結構9、產物結構13的分子結構Fig.6 Top view of molecular geometries of complex 11,transition state 9 and product 13
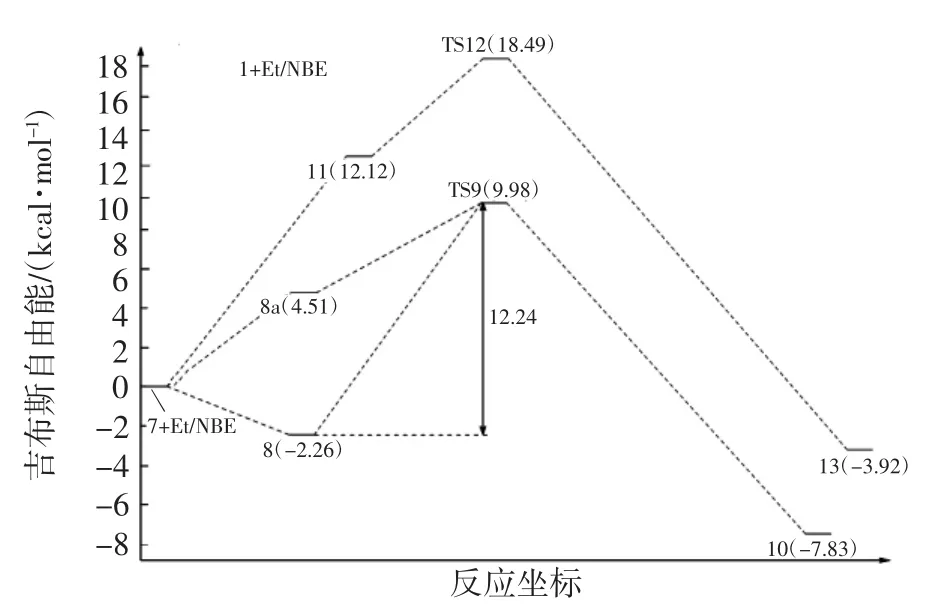
圖7 降冰片烯和乙烯再插入反應勢能圖Fig.7 Calculated potential energy diagram for norbornene and ethylene insertion into complex 7
對于乙烯單體的再插入反應,首先是乙烯單體對結構7進行配位,由于結構7的空間不對稱性,乙烯單體可從結構7后側或是前側進行配位,因此,本文模擬2種進攻方式的結構,并計算其能量.經過單點能計算出圖5中配合物結構8a(單體后側進攻)的勢能為4.51 kcal/mol,配合物結構8的勢能為-2.26 kcal/mol.對比2種進攻方式的勢能發現,中間體結構8a處于勢能淺井,比8具有更高的勢能,如圖7所示.因此,中間體結構8更為穩定,將優于結構8a率先形成.隨著中間體結構8的形成,乙烯再插入反應將通過過渡態結構9(TS9)生成最終產物結構10.觀察乙烯單體插入Ni—CH3鍵反應過程和插入Ni—NBE鍵反應過程,發現初始插入反應的過渡態3(TS3)與再插入反應的過渡態9(TS9)在結構上非常相似,如圖2和圖5所示.乙烯再插入反應過程的過渡態勢能為9.98 kcal/mol,比乙烯初始插入反應的過渡態勢能高2.58 kcal/mol.
對于降冰片烯插入Ni-NBE鍵反應,首先是降冰片烯單體與結構7進行配位,形成配位中間體結構11,然后通過過渡狀態12(TS12)獲得產物結構13,如圖6所示.與此同時可發現,降冰片烯再插入過程的過渡態結構12(TS12,圖6)和初始插入反應的過渡態結構6(TS6,圖3)也是非常相似的.
單點能計算結果表明,降冰片烯再插入過程的中間體結構11的勢能是12.12 kcal/mol,而乙烯再插入過程的中間體結構8的勢能是-2.26 kcal/mol,這表明乙烯比降冰片烯更容易與Ni—NBE結構進行配位.降冰片烯再插入反應的總能壘為18.49 kcal/mol,也遠高于乙烯單體再插入反應的總能壘(9.98 kcal/mol).因此,在體系中乙烯的再插入反應將比降冰片烯的再插入反應更容易發生.相關研究[16]也報道稱,由于降冰片烯的自身空間位阻,其連續插入反應是很難發生的.因此,單體再插入Ni—NBE鍵反應過程中,乙烯單體比降冰片烯更容易完成插入反應,所以結構10將是此階段主要產物結構.
2.4 乙烯的β-氫轉移反應和線性鏈增長反應的對比
乙烯線性鏈增長反應和β-氫轉移反應的理論計算研究[37-38]結果如圖8—圖10所示.
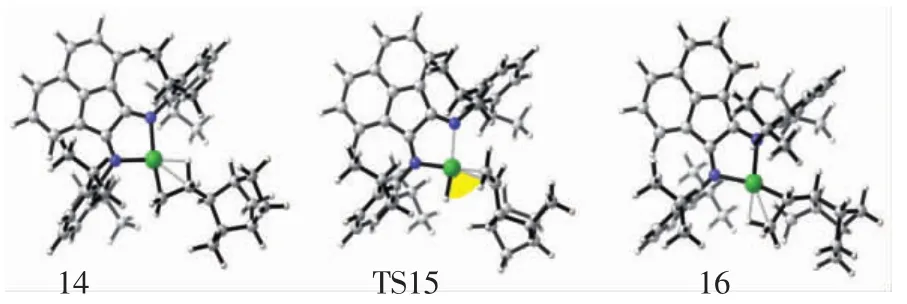
圖8 配合物結構14、過渡態結構15、產物結構16的分子結構示意圖Fig.8 Top view of molecular geometries of π-complex 14,transition state 15 and product 16
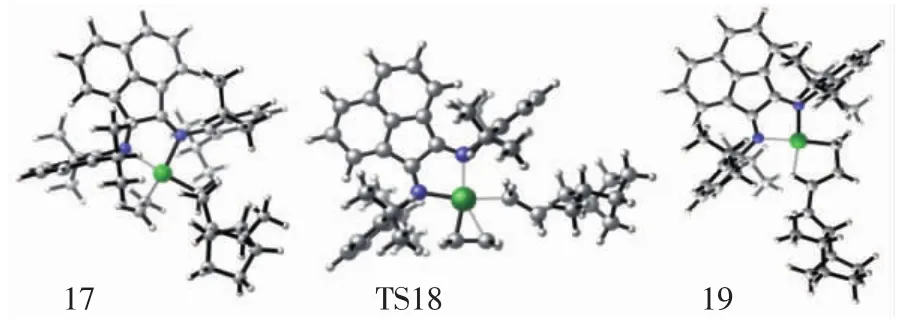
圖9 配合物結構17、過渡態結構18、產物結構19的分子結構Fig.9 Top view of molecular geometries of π-complex 17,transition state 18 and product 19
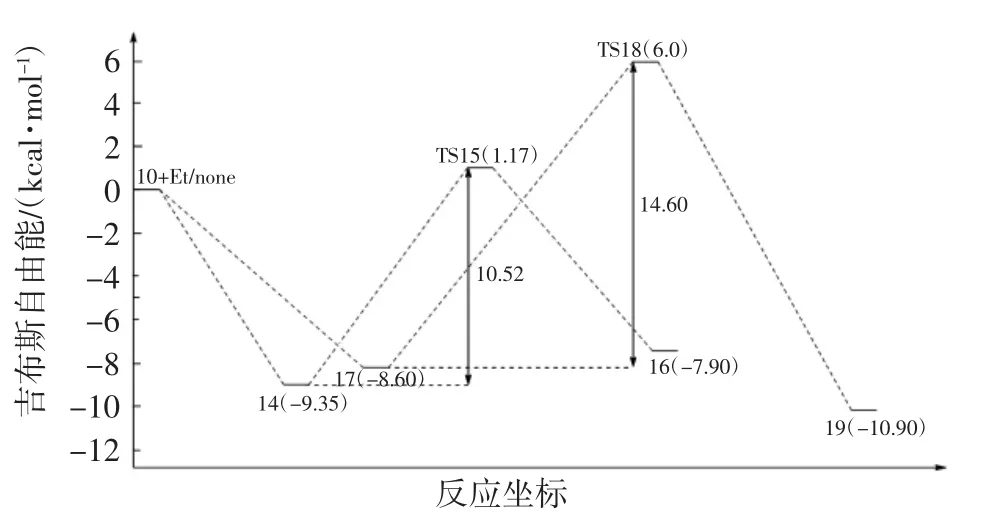
圖10 乙烯線性增長反應和β氫轉移反應的勢能圖Fig.10 Calculated potential energy diagram for β-hydride transfer reaction between linear propagation
對于乙烯β-氫轉移反應,其過渡態是非常難以尋找的.但根據文獻[38]的研究結果,構建出中間體結構15,該結構15與β-氫轉移反應過程的過渡態結構在能量和空間構型上是非常相似的.因此,本文把中間體結構15作為β-氫轉移反應的過渡態,如圖8所示.該反應從結構10開始,經歷結構14(β-agostic),然后通過過渡態結構 15(TS15)形成結構 16(hydride-olefin).中間體結構14的勢能是-9.35 kcal/mol,β-氫轉移反應的總能壘是1.17 kcal/mol,如圖10所示.
對于乙烯線性鏈增長過程,乙烯單體首先與結構10配位形成π復合物結構17,該配位結構與乙烯插入Ni—CH3過程中間體結構2也是相似的.隨著π復合物17的形成,鏈增長反應通過過渡態結構18形成產物結構19,如圖9所示.通過單點能計算表明,π配合物結構17的勢能是-8.6 kcal/mol,乙烯鏈增長過程的總能壘為6.0 kcal/mol,如圖10所示.
比較上述2個反應過程,發現β-氫轉移反應中的絡合過程比乙烯線性鏈增長的配位過程更容易發生,且乙烯線性鏈增長反應過程的總能壘要遠高于β-氫轉移反應過程的總能壘.因此,在該階段乙烯的β-氫轉移反應將比乙烯線性鏈增長反應更容易發生.
2.5 乙烯/降冰片烯無規共聚過程分析
對于本文采用催化劑形成的陽離子活性中心,通過計算乙烯/降冰片烯無規共聚過程中所有可能發生的主要反應,研究發現降冰片烯單體將比乙烯更容易發生插入反應,而在隨后的再插入過程中,由于降冰片烯單體自身的大空間位阻的影響,其連續插入是十分困難的,因此,再插入過程中乙烯將更容易發生反應.隨后,在乙烯不斷增長過程中,β-氫轉移反應將導致鏈段斷開,且轉移過程比線性鏈增長反應更容易發生.觀察β-氫轉移反應的終止結構與陽離子活性中心結構,發現兩者具有的結構是十分相似的,因此,單體可以再次進行插入反應.綜上所述,降冰片烯插入Ni-CH3鍵的引發反應、乙烯的再插入反應和增長鏈的β-氫轉移反應將構成循環體系,如圖11所示.該循環體系將導致乙烯/降冰片烯無規共聚反應只產生短鏈分子而鮮有長鏈聚合物產生,從理論上解釋了催化劑A催化乙烯和降冰片烯無規共聚無產物的原因.
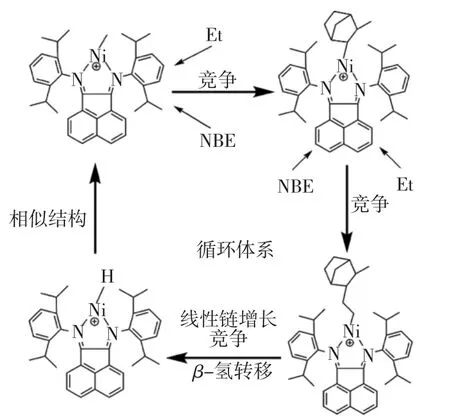
圖11 乙、降無規共聚過程的每步反應的平面結構Fig.11 Planar structure of each step in ethylene and norbornene copolymerization
3 結論
本文使用密度泛函數理論方法對N,N′-二(2,6-二異丙基)苊二亞胺溴化鎳催化的乙烯/降冰片烯無規共聚的反應過程進行研究,發現:在共聚反應中,降冰片烯單體將降低初始插入反應過程的能壘,優先乙烯插入陽離子活性中心,而乙烯將更容易發生隨后的再插入反應;但隨著鏈段增長,β-氫轉移反應將打斷鏈段的增長,導致該體系始終無法生成長鏈結構.通過理論計算,解釋了使用含有苊醌配體的二亞胺鎳化合物催化乙烯和降冰片烯無規共聚反應無聚合產物生成的原因.
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現代企業(2015年9期)2015-02-28 18:56:50
