抗炎中藥與阿爾茨海默病防治*
2019-01-18 11:04:40楊貴珍柯尊記
世界科學技術-中醫藥現代化 2019年9期
關鍵詞:中藥
楊貴珍,柯尊記
(1. 上海中醫藥大學基礎醫學院免疫學與病原生物學教研室 上海 201203;2. 上海中醫藥大學基礎醫學院神經科學研究中心 上海 201203)
阿爾茨海默病(Alzheimer's disease,AD)是一種年齡相關的神經系統退行性疾病,臨床表現為進行性的學習記憶能力減退,其病理特征是大腦中β 淀粉樣蛋白(Beta amyloid protein,Aβ)聚集形成的老年斑、tau蛋白過度磷酸化聚集形成的神經纖維纏結(Neurofibrous tangles,NFT)、長期慢性炎癥反應與膠質細胞增生、神經元死亡等。國際阿爾茨海默病組織(The Alzheimer's Disease International,ADI)估計到2030年,全世界癡呆患者人數將達到7500 萬人,2050 可能突破1.3 億人[1]。隨著人口老齡化步伐的加快,AD 已成為與心血管疾病、惡性腫瘤和中風并列的危害人類健康的“殺手”[2]。
1 神經系統炎癥對AD發生和發展的影響
對復雜的AD 的病理演變,研究者們提出了眾多假說來闡明。除了衰老這一自然因素之外,神經遞質紊亂、氧化應激、Aβ蛋白聚集、tau蛋白過度磷酸化、線粒體結構和功能異常、慢性炎癥等被認為是AD 的主要發病機制[3]。在慢性炎癥方面,研究者們就提出淋巴細胞、補體、激肽樣物質、氧自由基、細胞因子和趨化因子等多種參與因素(表1)。炎癥反應是當機體免疫系統面對病原體、衰老死亡細胞等異常信號產生的正向免疫應答,參與炎癥反應的免疫細胞、分子眾多,主要病理表現為血管通透性增加,炎癥細胞浸潤。研究表明,AD 患者神經系統炎癥反應明顯,是全身免疫調節紊亂的局部具體表現;腦內參與炎癥反應的細胞主要是神經膠質細胞[4]。神經系統炎癥在清除腦內衰老死亡細胞、突變細胞、抵抗病原體感染等方面發揮重要作用,但這些異常信號若長期存在,腦中膠質細胞將被過度激活,由此產生的免疫細胞因子、補體、激肽樣物質、氧自由基等在清除異物的同時,也會對腦神經元產生損害。這也是AD 腦內的主要病理學變化之一。本文綜述就神經系統炎癥的參與成分在AD 發生發展中的作用做簡要闡述。
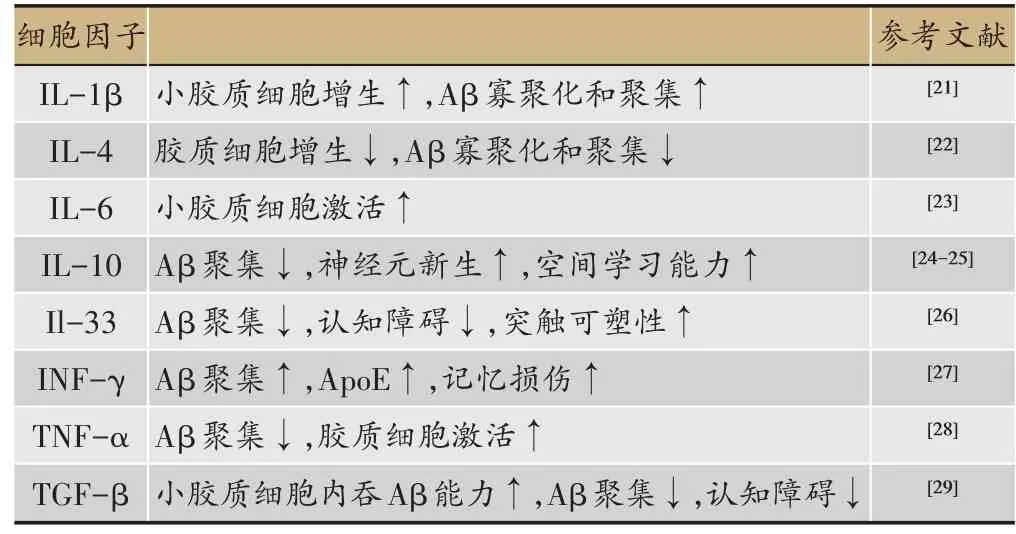
表1 與AD 發病機制相關的炎癥因子
1.1 小膠質細胞
小膠質細胞是腦內的巨噬細胞,能清除腦內衰老死亡的神經元和入侵的病原體。靜息狀態的小膠質細胞呈分枝狀,具有較強的吞噬能力和抗原提呈功能,也能分泌細胞因子、補體、神經營養因子來保護神經元。小膠質細胞低表達分化簇分子45(Cluster of differentiation 45,CD45)、主要組織相容性復合物Ⅱ(Major histocompatibility complex Ⅱ,MHCⅡ)等表面分子,吞噬能力較低[5]。活化狀態的小膠質細胞成阿米巴樣形狀,細胞表面模式識別受體如Toll樣受體、清道夫受體表達上調,合成和釋放大量的炎癥因子和氧自由基[6]。活化的小膠質細胞分泌的炎癥因子主要包括:腫瘤壞死因子-α(Tumor necrosis factor,TNF-α)、轉移生長因子-β(Transfer growth factor-β,TGF-β)、白細胞介素-1β(Interleukin-1β,IL-1β)、IL-6 及活性氧自由基(Reactive oxygen species,ROS)等。IL-1β 和IL-6 可激活神經元細胞內的淀粉樣前體蛋白(Amyloid Precursorprotein,APP)的分泌酶,促進APP的剪切,加速Aβ 蛋白的產生、沉積[7];IL-1β 能進一步活化膠質細胞并促進IL-6、TNF-α 及載脂蛋白-E4(Apolipoprotein-E4,Apo-E4)的分泌[8]。研究表明IL-1β 還可誘導tau 蛋白磷酸化,促進NFTs 的形成][9];TNF-α 濃度在AD 患者的血清、腦脊液以及Aβ 激活的膠質細胞中都可升高[10]。TNF-α濃度過高對神經元具有不利的影響,TNF-α 過表達的轉基因小鼠神經退行性病變更明顯,其機制可能是神經元上的TNF 受體(Tumor necrosis factor receptor,TNFR)與TNF-α 結合,介導caspase 級聯反應誘導神經元凋亡[11]。神經系統中存在TNFR-1 和TNFR-2 兩種TNFR,以不同的信號傳導通路保持腦內神經-免疫調節的平衡,AD 患者腦內出現神經網絡功能異常可能是這種平衡被打破了[12]。TGF-β 在AD 的腦損傷反應中也起著與上述炎癥因子類似的關鍵作用。AD 患者的大腦微血管高表達TGF-β通過促進血管內皮細胞釋放炎癥因子IL-1β和TNF-α,參與到AD炎癥的進程中[13]。ROS是機體內可以形成自由基的過氧化物以及參與氧代謝的含氧自由基,通過損壞神經元膜蛋白,造成細胞內外Ca2+穩態失衡,導致神經元凋亡。ROS 亦可導致自由脂酸大量釋放、前列腺素E2(Prostaglandin E2,PGE2)分泌增加以及Tau 蛋白的過度磷酸化,造成神經元的凋亡。NO 也是影響神經炎癥的重要介質,它通過產生對神經元膜蛋白毒性更強的過氧亞硝酸鹽起作用;一方面NO 能夠抑制線粒體呼吸鏈功能,干擾線粒體合成ATP ,最終導致神經元細胞能量耗竭而死亡;另一方面NO還能活化前列腺素合成酶,促進PGE2分泌。
在AD 病人腦內,激活的小膠質細胞與Aβ 斑塊共定位,小膠質細胞是腦內清除Aβ的主要細胞[14]。APP/PS1 轉基因小鼠腦內小膠質細胞降解吞噬的Aβ 的能力下降[15]。小膠質細胞激活在AD 發生發展過程的不同時期發揮不同的作用[16]。
1.2 星形膠質細胞
星形膠質細胞的主要作用是參與能量物質和神經遞質代謝,增強突觸可塑性等。AD 病程初期,星形膠質細胞通過分泌血管內皮生長因子,誘導血管形成,在Aβ 清除和分解中有重要作用[17]。當Aβ 聚集增加,激活的星形膠質細胞產生的NO 量增加,血管通透性增加,破壞血腦屏障(Blood brain barrier,BBB)的完整性[18]。AD 病程晚期,星形膠質細胞激活更加明顯,激活的星形膠質細胞表達鈣依賴磷酸酶(Calcineurin,CN)水平增加,CN 通過活化T 細胞核因子(Nuclear factors activating T cells,NFAT),引發包括突觸損失和/或可塑性改變,神經元凋亡等一系列病理學效應。在APP/PS1 小鼠,CN-NFAT 信號通路異常激活[19]。有AD 患者血漿和腦內的CD40 和CD40L 的水平升高,CD40-CD40L 相互作用可增加Aβ 生成,tau 蛋白過度磷酸化,而激活的星形膠質細胞是腦內CD40L 的主要來源[20]。
1.3 其它免疫細胞
B 細胞是體內的抗體產生細胞,B 淋巴細胞表面的CD40與T淋巴細胞表面的CD40L相互作用,對B淋巴細胞的增殖分化和激活起著關鍵性的作用。AD 患者血清中存在腦反應性抗體,包括天然抗體和自身抗體,這些抗體的病理生理意義還不清楚[30]。肥大細胞是組胺、花生四烯酸等增加血管通透性物質的主要來源。AD 患者腦內的肥大細胞明顯被激活,腦內約50%的組胺由肥大細胞分泌[31],聚集在BBB 周圍的肥大細胞還能分泌TNF-α、IL-1β、IL-8、IL33、CCL2、VEGF、ROS、PGE2等介質,參與BBB的通透性調節[32]。
1.4 補體系統
補體系統是固有免疫的重要組成部分,由補體(Complement C)C1 至C9 固有組分、調節組分及其受體組成。補體成分分泌增加及異常激活亦能加劇炎癥反應,誘導細胞死亡。補體系統也參與AD 發生和發展,纖維狀Aβ 可直接與C1q 的球部、C3 結合,通過經典通路和旁路通路激活補體系統,形成的復合物參與對衰老死亡的神經元清除作用[33]。同時生成的補體降解片段C3a、C5a起到激肽樣物質、過敏毒素的作用,能增加BBB 和血管的通透性,趨化免疫細胞進入腦組織中,加重神經炎癥反應對神經元的損傷。神經元因其低水平表達補體調節物(Complement regulator,CRegs),對補體介導的死亡特別敏感,AD 患者中神經元CRegs 表達上調[34]。C3 是體內含量最高的補體成分,其降解片段C3a 可以通過與C3aR 結合,增強吞噬細胞的吞噬能力,清除腦內沉積的的Aβ 斑塊與過度磷酸化的tau 蛋白纖維纏結,促進神經元再生;同時也能激活膠質細胞產生炎癥因子導致組織損傷。補體正常功能的發揮依賴于各組分之間相互作用的平衡,而這種平衡被打破在AD 發病發展中可能起重要作用[35]。小膠質細胞內NF-κB 信號通路激活后,可促進C3生成,與神經元上的C3aR 結合,導致神經元形態和功能損傷,而抑制C3a R 可改善AD 模型小鼠的學習認知障礙[36]。
2 基于炎癥假說的AD治療
目前,臨床上可用的治療AD 的藥物只有基于神經遞質假說的藥物,包括,利凡斯的明(rivastignine)、多奈哌齊(donepezil)、加蘭他敏(galantamine)和美金剛(memantine)。基于炎癥反應在AD 發生發展中的作用,人們試圖通過抑制炎癥過程來防治AD。目前正在進行的基于炎癥假說AD臨床試驗有近10 項。
2.1 非甾體類抗炎藥
在研究抗炎藥物防治AD 的過程中,非甾體類抗炎藥(Nonsteroidal antiinflammatory drugs,NSAIDs)表現出延緩和治療AD的潛力,其中有5項非甾體類抗炎藥,分別是氟比洛芬、布洛芬、羅非昔布、萘普生和塞來昔布正在進行臨床試驗。NSAIDs 被認為可抑制環加氧酶2(Cyclo-oxygenase-2,COX-2)的活性,減少PGE2 的生成,進而減輕炎癥反應。NSAIDs 雖能緩解炎癥癥狀、延緩病情惡化,長期服用也存在著消化系統、血液系統以及泌尿系統的嚴重副作用,影響了其臨床使用。
2.2 中藥防治AD
中國傳統醫學對AD 沒有統一的歸類,古代醫書中有“文癡”、“善忘”、“癲疾”等各種稱謂。在《左傳》中記載有“不慧,蓋世所謂白癡”、《華佗神醫秘傳》和《景岳全書》中開始稱為“癡呆”。中醫各個學派對AD病因病機歸納總結為腎虧髓虛、五臟不足為本,血瘀痰阻為標[37]。五臟六腑理論認為肝[38]、膽[39]、心、脾[40]均與AD 進展密切相關。經絡腧穴理論從經絡角度辨證論治,也取得了一定的臨床療效[41]。辨證論治是中醫治療的精髓,中藥對AD 的治療,主要從病因病機入手,采取中藥提取物、經典復方等多種方法,取長補短,融會貫通,長期進行臨床實踐及實驗研究。中藥復方因多靶點作用特點,具有整體治療的優勢。中藥提取物和中藥單體,因化學結構穩定、作用靶點明確、便于推廣等優勢,也受到研究者的青睞(表3)。
目前,運用中藥對神經炎癥性疾病進行治療已逐漸成為研究的熱點。近年來發現一些傳統中藥如人參、遠志、菖蒲、含笑、淫羊藿、雷公藤、厚樸、肉桂、穿心蓮、假馬齒莧、茱萸、銀杏等能改善AD 模型小鼠的認知障礙,這些藥物潛在的有效成分是開發治療AD新藥的新方向(表2)[42]。人們通過分離技術對這些藥用植物進行了提取,純化,有效成分分析檢測,發現多萜類、糖類、黃酮類、生物堿類化合物具有良好的抗神經炎癥的活性。中藥復方由于具有多靶點作用的特點,對于發病機制復雜的AD 治療具有其獨特優勢。當歸芍藥散、聰明湯和定志丸等中藥復方已體現出其防治AD的可能。
2.2.1 中藥防治AD的機制
人們對于這些中藥提取物和中藥單體抗神經炎癥的機制進行了廣泛的研究,發現其主要通過調節以下幾個方面起作用。首先,免疫調節作用。中藥有效成分對于外周組織和血液中淋巴細胞活性進行廣泛的調節,抑制血管活性物質和炎癥細胞趨化因子的釋放。白藜蘆醇、沒食子酸、雷公藤甲素等均能抑制肥大細胞的活化,減少組胺、白三烯等的分泌。黃芪皂苷Ⅳ體內、體外均可促進T、B 淋巴細胞的增殖,抑制小鼠腹腔巨噬細胞的活化[72]。腦內免疫調節作用對象主要是體現在抑制小膠質細胞過度激活上,假馬齒莧皂苷、穿心蓮內酯等中藥單均能抑制小膠質細胞的活性而減少小鼠腦內Aβ 斑塊的生成。部分中藥還能通過調節補體系統來抑制神經炎癥[73]。β-甘草次酸能調節C2、C3 的分泌,甘草酸苷能調控C3 分解成C3a,而C3a 是體內重要的過敏毒素[74]。羽扇豆三萜醇能抑制C3 轉化酶活性產生抗炎效果[75]。其次,抑制炎癥信號通路發揮作用。第一,通過阻斷NF-κB 信號通路起效。NF-κB 信號通路是機體內最重要的參與炎癥反應的通路,在衰老因素的誘導沉積在腦內的Aβ 斑塊可激活NF-κB 信號通路,導致核轉錄因子磷酸化激活后能夠進入細胞核內增強炎癥因子基因的表達。中藥提取物通過抑制NF-κB 的活化則能夠有效抑制膠質細胞激活,從而發揮抗神經炎癥作用。第二,通過阻斷MAPK 信號通路起效。沉積在腦內的Aβ 斑塊亦可激活絲裂原活化蛋白激酶,導致核轉錄因子磷酸化激活后能夠進入細胞核內增強炎癥因子基因及炎性蛋白iNOS 及COX-2 的表達[76]。中藥提取物和中藥單體通過抑制NF-κB 和MAPK 信號通路而對AD 的防治產生很好的效果。第三,通過阻斷PI3K/AKT 信號通路起效。PI3K/AKT信號通路在體內作用廣泛,此信號通路激活也參與了神經炎癥的發生發展。最后,通過清除活性氧自由基[77]起作用。

表2 靶向炎癥因子治療AD相關中藥研究

表3 用于AD防治的中藥復方
3 小結
綜上所述,神經炎癥在AD 的發生發展過程中發揮重要作用。外周的淋巴細胞、補體系統、激活的小膠質細胞及炎癥因子等在AD 炎癥反應過程中扮演著重要的角色。NSAIDs可以減緩AD的發展,NSAIDs的副作用影響了其在臨床上使用。雖然基于炎癥假說的AD 治療藥物已有諸多失敗案例,但現在仍在進行中的臨床試驗還存在一線希望。尋找安全有效的AD治療方案可能需要綜合Aβ假說、tau假說、神經元保護假說以及炎癥假說。AD 的病因不是單一機制造成的,從多靶點作用特點的中藥中有望找到防治AD 的潛在藥物。
猜你喜歡
中老年保健(2021年5期)2021-12-02 15:48:21
中老年保健(2021年4期)2021-12-01 11:19:40
中老年保健(2021年4期)2021-08-22 07:08:32
中國現代中藥(2020年10期)2020-12-16 08:53:18
金橋(2020年7期)2020-08-13 03:07:00
基層中醫藥(2020年12期)2020-07-22 06:34:38
中國現代中藥(2020年4期)2020-06-10 09:56:34
基層中醫藥(2018年6期)2018-08-29 01:20:20
長春中醫藥大學學報(2017年1期)2017-04-16 05:56:49
肝博士(2015年2期)2015-02-27 10:49:49
