氣相色譜法同時測定乳制品中有機氯農藥和多氯聯苯殘留量的研究
2018-12-29 03:42:50李霞李潔李玲王艷麗申中蘭劉艷明祝建華
中國乳品工業 2018年11期
李霞,李潔,李玲,王艷麗,申中蘭,劉艷明,祝建華
(山東省食品藥品檢驗研究院濟南 250101)
0 引 言
乳制品中藥物殘留和環境污染的問題已經拉響我國奶源安全的警報。多氯聯苯和有機氯農藥是國際環境科學領域十分關注的持久性有機污染物。此類化合物具有親脂性和半揮發性,難以被生物降解,可通過食物、水、大氣和土壤等環境介質與包括人類在內的環境生物體系接觸,對生態環境和人類的健康帶來潛在的危害,因此建立多氯聯苯和有機氯農藥殘留的檢測方法對于保證乳制品的質量十分重要。
目前,多氯聯苯和有機氯農藥的測定對象主要有水[1-3],土壤[4],水產品[5-7],蛋及蛋制品[8],農殘品[9],乳制品相對較少。多氯聯苯和有機氯農藥的檢測方法有氣相色譜法,氣相色譜-質譜聯用法[10]。乳制品中有機氯殘留常用的凈化技術有固相萃取法[11]、凝膠滲透色譜凈化[12]、酸化法等,其中乳制品中有機氯殘留的測定采用磺化法的報道也不多[13],乳制品中多氯聯苯的測定有采用索氏提取法及自動凈化處理法[14]。至今濃硫酸磺化法作為同時檢測乳制品中的多氯聯苯和有機氯農藥殘留的前處理方法尚鮮見報道。本文應用超聲提取法同時提取乳制品中的多氯聯苯和有機氯農藥殘留,采用濃硫酸凈化的前處理方法,提取方法簡便,濃硫酸磺化法凈化效果好,回收率高,色譜背景干凈,即快速又節約成本。同時也參考歐盟、日本農藥殘留的檢測方法[15-16]和國內的標準[17-18]。所以本研究對于同時測定乳制品中有機氯和多氯聯苯殘留量的檢測有很好的借鑒作用。
l 實驗
1.1 主要儀器與試劑
氣相色譜儀:Agilent 6890型,配有電子捕獲檢測器(FID),美國Agilent公司;旋轉蒸發器:Buchi R-200型,瑞士Buchi公司;7種指示性多氯聯苯混標(內含PCB28、PCB52、PCBl01、PCBll8、PCBl38、PCBl53、PCBl80,10μg/mL,純度99.0%,溶于環己烷),德國Dr.Ehrenstorfer公司;ɑ-666、β-666、γ-666、δ-666、p,p’-DDE、o,p’-DDD、p,p’-DDD及p,p’-DDT,1 000μg/mL,純度均大于99%,中國農業部環境保護監測所;七氯,1 000μg/mL,純度均大于99%,中國農業部環境保護監測所;艾氏劑,1 000μg/mL,純度均大于99%中國農業部環境保護監測所;無水硫酸鈉:分析純,于600℃下干燥4 h,冷卻后儲存于干燥的密閉容器中,備用;氯化鈉:分析純;乙腈:優級純;實驗所用其它試劑均為分析純;實驗用水為二次蒸餾水。
1.2 色譜條件
色譜柱:HP-1701型石英毛細管柱(30 m×0.25 mm×0.25μm);色譜柱升溫程序:50℃保持1 min,20℃/min升至200℃保持1 min,5℃/min升至230℃保持5 min,5℃/min升至260℃保持10 min;進樣口溫度:290℃;檢測器溫度:300℃;載氣、輔助氣:均為氮氣,純度為99.999%,載氣流速為3 mL/min;進樣方式:不分流進樣;進樣量:1μL。
1.3 標準溶液的配制
分別稱取適量各種多氯聯苯、六六六、滴滴涕,七氯,艾氏劑,用正己烷配制成0.5μg/mL儲備液。根據需要,用正己烷再配制成所需濃度的混合標準溶液。
1.4 試驗方法
液體樣品稱取2.0 g樣品于50 mL離心管中,加入2 g氯化鈉,加入20 mL乙腈提取液,渦旋2 min,超聲30 min,再加入無水硫酸鈉5 g脫水,渦旋2 min,于8 000 r/min離心3 min,將上清液轉移到50 mL旋轉蒸發瓶中,下層樣品再提取一次后合并上清液,40℃水浴中旋轉蒸至近干,加入10 mL正己烷復溶,再加入1 mL硫酸磺化,于8 000 r/min離心3 min,后取上清液氮氣吹干,準確加入1 mL正己烷復溶,再加入0.2 mL濃硫酸磺化,8 000 r/min離心3 min,取上清液用純凈水洗至中性,取上清液進入氣相色譜分析。
固體樣品稱取2.0 g樣品于50 mL離心管中,加入10 mL水,渦旋0.5 min,加入2 g氯化鈉,再加入20 mL乙腈提取液,渦旋2 min,超聲30 min,放入離心機,8 000 r/min離心3 min,取上清液于50 mL離心管中,加入5 g無水硫酸鈉脫水,轉移到50 mL旋轉蒸發瓶中,下層樣品再提取一次后合并上清液,40℃水浴中旋轉蒸至近干,加入10 mL正己烷復溶,加入1 mL硫酸磺化,于8 000 r/min離心3 min,后取上清液氮氣吹干,準確加入1 mL正己烷復溶,再加入0.2 mL濃硫酸磺化,8 000 r/min離心3 min,取上清液用純凈水洗至中性,取上清液進入氣相色譜分析。
2 結果與討論
2.1 提取條件的優化
2.1.1 提取溶劑的選擇
六六六、滴滴涕、七氯、狄試劑和多氯聯苯屬于極性物質,乳制品中脂類物質是非極性物質,根據相似相容原理,選用極性有機溶劑提取目標物可以減少脂類物質的干擾。實驗對乙腈、甲醇兩種極性提取溶劑進行考察。結果表明:乙腈作為提取溶劑時,乳制品中4種六六六、4種滴滴涕、七氯、狄試劑和7種多氯聯苯的加標回收率比甲醇作為提取溶劑時得到的加標回收率好,所以選用乙腈為本實驗的最佳提取溶劑。
2.1.2 提取方法
提取方法的選擇實驗以鮮牛奶樣品的加標回收率來考察超聲提取法,分別以超聲10 min,20 min,30 min,40 min進行考察。17種化合物的加標回收率如圖1所示。超聲10 min的加標回收率在70.58%~81.68%,超聲20 min的加標回收率在75.63%~88.23%,超聲30 min的加標回收率在83.55%~95.38%,超聲40 min加標回收率在83.6%~95.57%,超聲30 min和超聲40 min兩者相差不大,考慮到試驗時間的節約,所以選擇超聲30 min提取法的最佳超聲時間。

圖1 不同超聲時間的回收率
2.1.3 凈化條件的優化
以鮮牛奶樣品為對象來考察濃硫酸用量,分別加入量為0.5、1.0、2.0 mL的濃硫酸進行考察,不同用量濃硫酸對空白鮮牛奶樣品的凈化效果見圖2。實驗結果表明:在加入0.5 mL濃硫酸后,經渦旋、離心,提取物溶液發生分層但上層溶液仍存在一些黃色的脂類物質,而當加入1.0 mL和2.0 mL的濃硫酸時,提取物溶液發生分層明顯且上清液澄清,有較好的凈化作用,同時考慮溶劑的節約,所以選擇濃硫酸的用量為1.0 mL。加標量為10μg/kg的鮮牛奶、酸牛奶和奶粉的凈化效果如圖3。在實驗過程中,單獨使用濃硫酸凈化后會產生離子型雜質及濃硫酸的殘留,這些物質會污染氣相色譜儀的毛細管柱,且目標物有干擾峰出現,而加蒸餾水洗至中性后對氣相色譜儀的毛細管柱起到保護作用,并且化合物的回收率不受影響。加蒸餾水水洗和不加水洗的效果圖如圖4。
2.1.4 色譜柱的選擇
本實驗在確定進樣口溫度、檢測器溫度、載氣流速及其他相關條件一致的情況下,實驗中采用HP-5(30 m×0.25 mm×0.25μm)柱子無法使其中的某些化合物分開。經過多次試驗,試驗中采用DB-1701(30 m×0.25 mm×0.25μm)的色譜柱可以使所有化合物達到很好的分離,并且在保證分離效果的前提下,改變升溫程序還可以縮短分析樣品所需時間。所得色譜最佳分離條件如前所述,色譜分離效果見圖2。
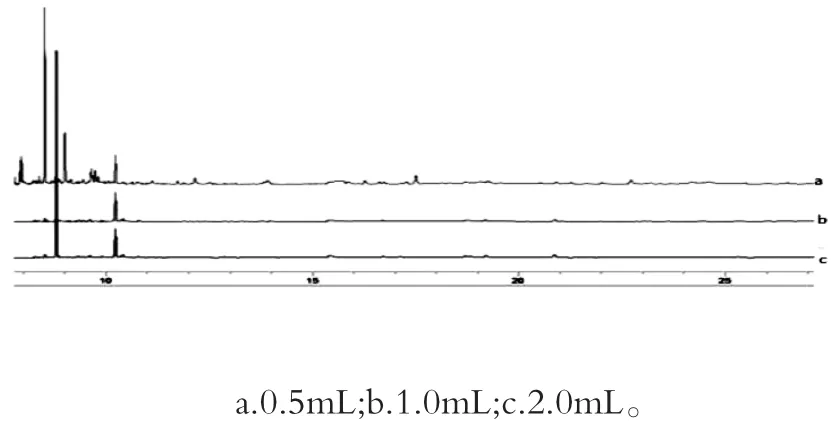
圖2 不同濃硫酸加入量對空白鮮牛奶樣品的凈化效果

圖3 加標量(10μg/kg)鮮牛奶、酸牛奶及奶粉的的凈化效果

圖4 加標量(10μg/kg)鮮牛奶加蒸餾水水洗及不加蒸餾水水洗的效果
2.2 方法的考察
2.2.1 線性范圍、檢出限和定量限
實驗以空白鮮牛奶樣品為基質,各稱取鮮牛奶2.0 g于一系列50 mL具塞離心管中,1.4節的方法進行前處理得到空白樣品基質,依次加入待測組分得到質量濃度為5、10、20、40、100、200μg/L的一系列基質標準溶液,按1.2節的分析方法測定,采用外標法定量。以峰面積為縱坐標(y),目標物的質量濃度為橫坐標(x,μg/L)繪制基質標準曲線,線性范圍結果見表1。方法的檢出限和定量限范圍分別為0.08~0.27μg/kg和0.27~0.90μg/kg,可滿足乳制品中4種六六六、4種滴滴涕、七氯、艾氏劑和7種指示性多氯聯苯殘留分析的要求。實驗中以所得到的3倍信噪比(RSN=3)計算檢出限,以所得到的10倍信噪比(RSN=10)計算定量限,檢出限和定量限的結果見表1。

表1 4種六六六、4種滴滴涕、七氯、艾氏劑和7種指示性多氯聯苯的線性范圍、線性方程、相關系數(r)、檢出限
2.2.2 加標回收率
實驗分別加標量為5、10、50 mg/kg的鮮牛奶、奶粉和酸奶(每個水平重復6次),按照1.2節和1.4節的方法進行前處理和分析測定,計算加標回收率和RSD,結果見表2-4表。

表2 純牛奶中添加4種六六六、4種滴滴涕、七氯、艾氏劑和7種多氧聯苯的加標回收率和RSD(n=6)

表3 酸奶中添加4種六六六、4種滴滴涕、七氯、艾氏劑和7種多氧聯苯的加標回收率和RSD(n=6)
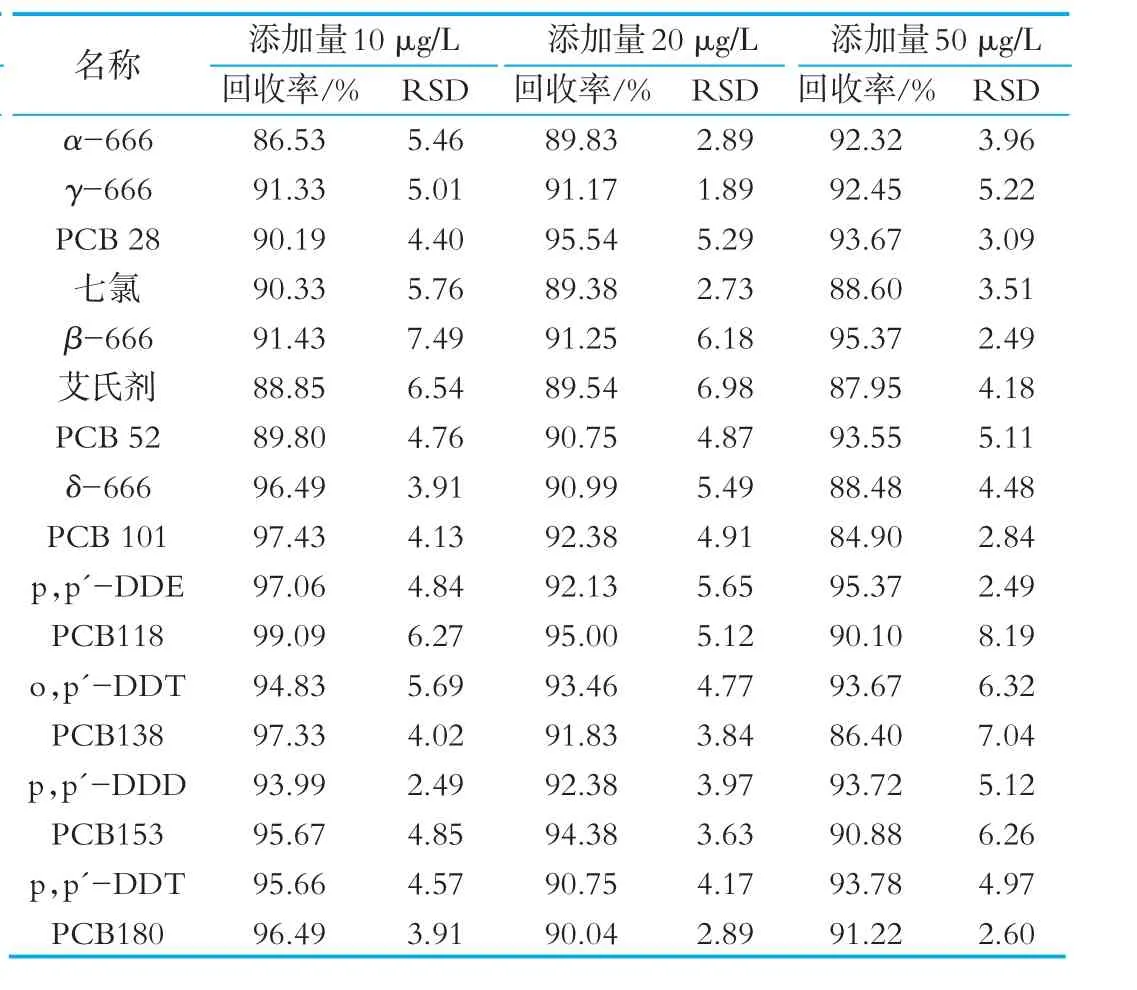
表4 奶粉中添加4種六六六、4種滴滴涕、七氯、艾氏劑和7種性多氧聯苯的加標回收率和RSD(n=6)
表2-表4表明:在10、20、50 mg/kg 3個加標水平下,純牛奶空白樣品中17種化合物的加標回收率范圍為87.13%~99.40%,RSD范圍為2.17%~7.67%;酸奶空白樣品中17種化合物的加標回收率范圍為85.09%~97.65%,RSD范圍為1.23%~8.12%;奶粉空白樣品中17種化合物的加標回收率范圍為86.40%~97.93%,RSD范圍為1.89%~7.04%。該方法的準確度與精密度高,符合鮮牛奶、酸奶和奶粉中4種六六六、4種滴滴涕、七氯、艾氏劑和7種指示性多氯聯苯殘留分析的要求。
2.2.3 定量方法的選擇
實驗表明:使用溶劑標準曲線對乳制品樣品中4種六六六、4種滴滴涕、七氯、艾氏劑和7種多氯聯苯進行定量所得到的加標回收率均超出110%,這是因為在氣相色譜分析中這些化合物表現出的不同程度的基質增強效應,使得待測物檢測信號增強。為了提高方法的可靠性和結果的準確度,消除基質效應,在實驗室中采用不含待測物的空白樣品經過1.4節方法進行處理,加入與溶劑標準曲線相同系列濃度的待測組分,即制作基質標準曲線來進行定量分析。實驗中分別對純牛奶,酸牛奶和奶粉配制了基質標準曲線,結果表明,三種樣品空白基質曲線中的待測組分的響應值和標準曲線的斜率基本相同,所以為了避免每個樣品種類都要配制其基質匹配標準曲線,實驗中選取相對雜質較少的鮮牛奶空白基質作基質曲線。實驗表明使用基質標準曲線進行定量得到的加標回收率在80%~110%之間,消除了基質效應的影響,同時提高方法的可靠性和結果的準確度。
2.2.4 實際樣品的檢測
實驗中對隨機送檢的30批乳制品分別按照1.2節和1.4節的方法進行前處理和分析測定,結果表明:純牛奶,酸奶和奶粉中的4種六六六、4種滴滴涕、七氯,艾氏劑和7種指示性多氯聯苯均未檢出。
3 結論
本實驗建立了濃硫酸凈化的前處理方法,在乳制品六六六、滴滴涕,七氯,艾氏劑和指示性多氯聯苯的檢測中取得較好的凈化效果,并且結合氣相色譜法實現對乳制品中六六六、滴滴涕,七氯,艾氏劑和指示性多氯聯苯的同時定量分析,適用于乳制品中六六六、滴滴涕,七氯,艾氏劑和指示性多氯聯苯的快速檢測。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
中國乳業(2018年3期)2018-04-13 01:05:08
食品與機械(2017年4期)2017-07-05 14:46:17
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
現代食品(2016年24期)2016-04-28 08:12:06
工業設計(2016年12期)2016-04-16 02:51:53
食品安全導刊(2011年6期)2011-04-12 00:00:00
