微波消解-原子吸收光譜法測定羧基麥芽糖鐵中鉛和砷
2018-12-14 12:30:50朱曉蕾張厚森
食品與藥品 2018年6期
唐 健,朱曉蕾,張厚森*
(江蘇省理化測試中心,江蘇 南京 210042)
羧基麥芽糖鐵是治療缺鐵性貧血(iron deficiency anemia,IDA)的新型第三代鐵復合物。它可有效補充人體內的鐵,滿足正常紅細胞生成的需要[1-5]。羧基麥芽糖鐵的鐵含量通常在20%以上,高者達40%[6-7]。鐵元素的存在對其他痕量元素的測定有很大的干擾。鉛、砷測定常用方法有原子熒光光譜法、原子吸收光譜法[8-11]、電感耦合等離子質譜法(ICP-MS)[12-15]等。原子熒光光譜法和原子吸收光譜法檢出限低、精密度好,非常適合痕量鉛、砷含量的測定。但原子熒光光譜法至今未能收入中國藥典[16];ICP-MS能多元素同時測定,檢出限與原子熒光光譜法和石墨爐原子吸收光譜法相當甚至略有優勢,但對于高鹽基樣品的測定濃度有限制,高濃度樣品會對截取錐和檢測器造成一定程度的影響[12]。同時ICP-MS昂貴的價格也影響了其通用性。本實驗中羧基麥芽糖鐵中的鐵含量為23%~27%,對測定干擾很大。采用微波消解前處理技術,基體匹配標準加入法,用氫化物發生原子吸收光譜法測定砷含量,用石墨爐原子吸收光譜法測定鉛含量,取得了良好的實驗結果。
1 儀器與試藥
1.1 儀器
AA240FS-GTA120型石墨爐原子吸收光譜儀(VGA-77氫化物發生器,美國瓦里安公司);TOPEX型微波消解儀(上海屹堯儀器公司);Astacus型超純水系統(德國 MembraPure公司);TT-6型不銹鋼控溫電熱板(南京同吉燦儀器儀表公司)。
1.2 試劑與藥品
鉛、砷標準溶液(質量濃度均為1000 mg/L,國家鋼鐵材料測試中心鋼鐵研究總院);硝酸(優級純,成都科隆);鹽酸(優級純,南京化學試劑公司);硼氫化鉀,氫氧化鉀,抗壞血酸,硫脲(分析純,國藥集團);羧基麥芽糖鐵原料藥(國內某制藥公司,批號:0160103,0160105,0160106,0170205,0170207,0170215)。
2 方法與結果
2.1 儀器工作條件
2.1.1 石墨爐原子吸收光譜儀工作條件 波長283.3 nm;燈電流:5.0 mA;狹縫:0.5 nm;干燥溫度:90~120 ℃,斜坡式升溫;灰化溫度:400 ℃,原子化溫度:2100 ℃;基體溶液加入量:10 μl。
2.1.2 氫化物-原子吸收光譜儀測定砷的工作條件 波長193.7 nm;燈電流:6.0 mA;狹縫:0.5 nm;空氣流量:13.50 L/min,乙炔流量:2.10 L/min;載氣為氬氣。
2.2 溶液制備
2.2.1 試劑配制 5%抗壞血酸-硫脲溶液:稱取抗壞血酸、硫脲各25 g于純水中,溶解稀釋至500 ml;0.3%硼氫化鉀溶液:稱取3.0 g硼氫化鉀,溶于氫氧化鉀溶液(5 g/L)中,并定容至1000 ml;5%鹽酸溶液(v/v):移取50 ml鹽酸于純水中,稀釋定容至1000 ml。
2.2.2 鉛含量測定樣品制備 稱取羧基麥芽糖鐵原料藥(批號:0160103)0.3 g(精確至0.0001 g)于微波消解罐中,加入硝酸5 ml和雙氧水2 ml,密封置于微波消解儀中,依照微波消解程序將樣品消解完全,放冷,純水多次洗滌消解罐,合并于燒杯中,在控溫電熱板上120 ℃加熱使多余的酸揮發除盡,待溶液揮發剩余約2~3 ml時,取下冷卻,純水定容至25.0 ml。同法制備不加樣品的試劑空白。微波消解條件見表1。
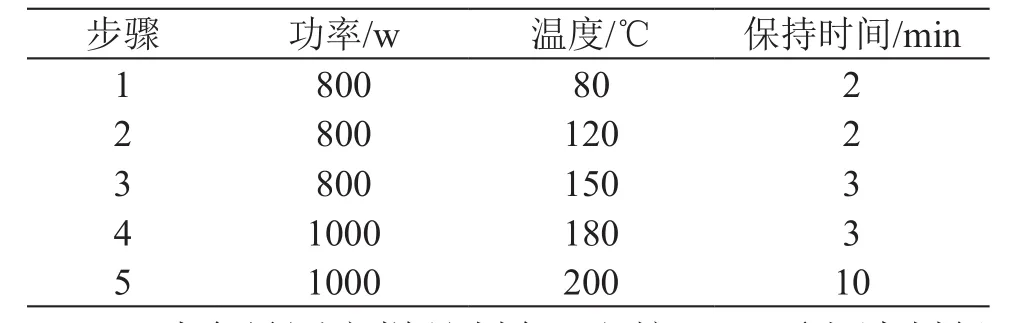
表1 微波消解程序
2.2.3 砷含量測定樣品制備 取按2.2.2項方法制得樣品溶液2.0 ml,加入5%抗壞血酸-硫脲溶液2 ml和鹽酸0.5 ml,純水稀釋至10.0 ml。同法制得砷空白溶液。基體溶液:取試樣4份,每份0.3 g(精確至0.0001 g),按2.2.2項下微波消解程序消解,全部合并洗脫在大燒杯中,在控溫電熱板上,120 ℃加熱使多余的酸揮發除盡,待溶液揮發剩余約3~5 ml時,取下冷卻,純水定容至100.0 ml。
2.2.4 標準曲線制備 精密量取鉛單元素標準溶液(1000 mg/L)適量,逐級稀釋配制成濃度為100 μg/L鉛標準溶液,作為母液;儀器自動稀釋制成標準曲線各濃度點。取2.2.2項下樣品溶液,自動進樣時設置與標準曲線各濃度點共同加入到石墨管中,加入量為10 μl,則制成含樣品溶液10 μl的濃度為5.0,10.0,20.0,40.0,50.0 μg/L的鉛標準系列。
精密量取砷元素標準溶液(1000 mg/L)適量,分別用5%鹽酸溶液逐級稀釋至濃度為0.1 mg/L的對照品儲備液。分別取0,0.5,1.0,2.0,4.0,5.0 ml對照品儲備液于6個50 ml量瓶中,加入5%抗壞血酸-硫脲溶液10 ml,2.2.3項下基體溶液10 mL,加5%鹽酸溶液稀釋至刻度,得濃度分別為0,1.0,2.0,4.0,8.0,10.0 μg/L系列標準溶液。以0.3%硼氫化鉀溶液和5%鹽酸溶液為載流溶液,按程序自動進樣于氫化物-原子吸收光譜儀中,依次測得砷標準系列。
2.3 線性試驗和方法檢出限
依法將配制好的系列標準溶液按2.1.1、2.1.2方法分別進樣測定,繪制標準曲線;并對測定數據進行線性回歸分析,得到線性方程。制備測定條件下的試劑空白溶液20份,測定,求其標準偏差(SD),以3倍SD作為檢出限(LOD),以10倍SD為定量限(LOQ)。同時,根據樣品稱樣量和定容體積計算方法檢出限。結果見表2。

表2 標準曲線及檢出限、定量限
2.4 精密度試驗
分別取含鉛濃度為20.0 μg/L和砷濃度為4.0 μg/L的標準溶液,連續測定6次,計算精密度RSD為3.41%(鉛),2.43%(砷)。結果表明儀器的精密度良好,見表3。

表3 精密度試驗結果
2.5 重復性試驗
取樣品0.3 g(精確至0.0001 g),按2.2.2和2.2.3項下方法消解,每種元素平行制備6份。按2.1.1 和2.1.2項條件測定,鉛含量0.87 mg/kg,RSD為3.91%,砷含量1.46 mg/kg,RSD為4.31%。結果表明,方法重復性良好,結果見表4。
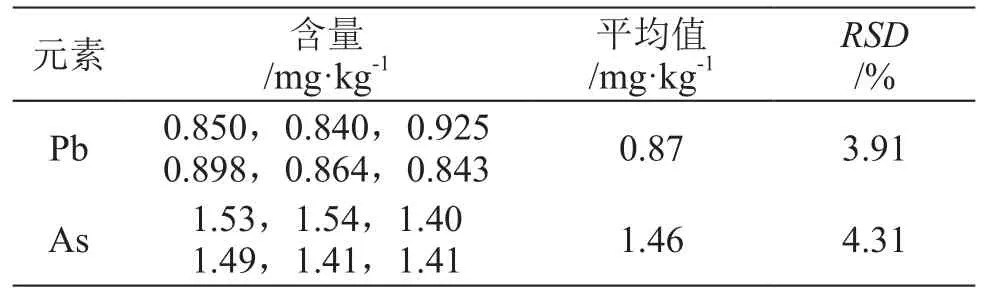
表4 重復性試驗結果
2.6 加標回收試驗
稱取同批號的羧基麥芽糖鐵原料藥0.3 g(精確至0.0001 g),每種元素各取3個濃度值作為加標回收試驗點,鉛加入濃度為10.0,20.0,30.0 μg/L,砷加入濃度為2.8,3.6,4.4 μg/L,每個濃度值平行制備3份樣品,按2.2.2和2.2.3項下方法消解,按2.1.1 和2.1.2項下條件測定。結果見表5。
2.7 樣品測定
取6個批號的樣品(0160103,0160105,0160106,0170205,0170207,0170215)各0.3 g,按規定方法制備樣品溶液,按2.1.1、2.1.2項條件測定,樣品中鉛、砷含量結果見表6。

表5 回收率試驗結果

表6 樣品測定結果
3 討論
3.1 標準加入法的選擇
分析結果的準確性直接依賴于標準樣品和未知樣品物理化學性質的相似性。標準系列與樣品基體的精確匹配是制備良好校正曲線的必要條件。試樣物理化學性質的變化會引起原子化效率、基體效應、背景和干擾情況的改變,導致測定誤差的增加。羧基麥芽糖鐵中鐵含量很高,嚴重干擾鉛和砷的測定,如采用普通標準曲線法,則標準曲線基體溶液和樣品的基體溶液嚴重不匹配,對靈敏度、精密度和回收率影響均很大。因此采用基體匹配標準加入法,可減少樣品基體的物理化學干擾,提高測定的準確度。
3.2 石墨爐基體改進劑的選擇
通過研究比較,發現采用0.5%磷酸二氫銨作為石墨爐的基體改進劑且加入量為共進2 μl,能有效克服背景干擾,增加穩定性。
3.3 石墨爐升溫程序的選擇
采用90~120 ℃斜坡式升溫的方式進行干燥,既避免了樣品溶液的沸騰和飛濺,又保證較快的蒸干速度。灰化溫度采用400 ℃的平臺溫度,保證了元素不損失的情況下最大化地破壞基體組分,除去樣品中的易揮發組分,減少基體干擾。原子化階段采用最大功率升溫,石墨爐以最快速度升到原子化溫度,獲得最高靈敏度。最大功率升溫具有靈敏度高、實際原子化溫度較低的特點,有延長石墨爐壽命的作用[17]。
3.4 硼氫化鉀的用量
硼氫化鉀的濃度對氫化物的形成影響很大,測定砷需要的硼氫化鉀濃度通常為0.6%,但樣品基體中含高濃度的鐵、銅和鎳時,使用0.3%硼氫化鉀可避免以上干擾,因此本文采用0.3%硼氫化鉀溶液。
3.5 酸介質及其用量
在經常使用的各種酸中,由于氫化物原子化器是石英材質,通常禁止使用含氫氟酸溶液。硫酸和高氯酸會降低或使信號喪失,且這兩種酸具有強氧化性,所以應盡量避免使用。鹽酸、硝酸均可作為載流生成氫化物,因此是常用的酸介質。但硝酸具有氧化性,會與加入的還原劑起氧化還原反應,削弱還原劑的作用,因此選擇鹽酸作為氫化物生成介質。試驗研究了鹽酸體積分數在1%~8%范圍內改變時對同一熒光度的影響,結果見圖1。由圖1可見,鹽酸體積分數為5%時吸光度最高;鹽酸體積分數低于5%時,吸光度逐漸降低,因此選擇鹽酸的體積分數為5%。

圖1 鹽酸的體積分數與砷標準溶液吸光度的關系
3.6 還原劑的選擇
5%抗壞血酸-硫脲混合溶液既是良好的還原劑,又是抗干擾的絡合劑[18]。它能在室溫條件下完全還原試樣溶液中的砷離子為可氣態的三價離子。溶液中大量存在的過渡元素會優先消耗硼氫化鉀,還原生成微細且分散的金屬沉淀,吸附或催化分解氫化物,強烈抑制砷的分析信號。加入5%抗壞血酸-硫脲混合溶液后,干擾嚴重的過渡金屬銅、鐵、鎳會在溶液中形成穩定的絡合物,不被還原成微細、分散的沉淀干擾測定,從而起到穩定溶液抗干擾的作用。
在樣品中鐵含量高達20%以上的測定干擾下,采用基體匹配標準加入法并優化測定條件,能很好地避免高鐵含量帶來的測定干擾,可準確測定樣品中的鉛、砷含量。
