高效液相色譜法測定復方銀翹氨敏膠囊中馬來酸氯苯那敏含量及含量均勻度
2018-11-20 07:42:36吳曉燕尚春燕
中國藥業 2018年22期
吳曉燕,唐 琨,尚春燕
(1.四川省樂山市食品藥品檢驗檢測中心,四川 樂山 614000; 2.樂山職業技術學院,四川 樂山 614000)
復方銀翹氨敏膠囊(曾用名:雙撲維C銀翹膠囊、維C銀翹膠囊)為常見抗感冒藥,是由對乙酰氨基酚、馬來酸氯苯那敏、維生素C及銀翹浸膏等組方的中西藥復方制劑,現行標準收載于《國家藥品標準·化學藥品地方標準上升國家標準(第四冊)》[1],但僅對維生素C和對乙酰氨基酚的含量進行了測定,對處方中具有較強抗組胺作用的馬來酸氯苯那敏并未規定明確的測定方法。本研究中采用高效液相色譜法對該品種中馬來酸氯苯那敏的含量及含量均勻度[2-7]進行了測定,現報道如下。
1 儀器與試藥
Agilent 1260型高效液相色譜儀,Agilent OpenLAB色譜工作站,二極管陣列檢測器(美國安捷倫科技有限公司);XS205 DualRange型電子天平(瑞士Mettler Toledo公司)。
馬來酸氯苯那敏對照品(中國食品藥品檢定研究院,批號為 100047-2015-07,按 C16H19ClN2·C4H4O4計,含量為99.7%);復方銀翹氨敏膠囊(委托企業,四川彩虹制藥有限公司,受托企業,四川恩威制藥有限公司,批號為 170311,170501,170314);甲醇為色譜純,磷酸二氫鉀、三乙胺為分析純,試驗用水為純化水。
2 方法與結果
2.1 色譜條件
色譜條件:Waters XTerra RP18柱(250 mm ×4.6 mm,5 μm);流動相:A 相為甲醇,B 相為 0.02 mol/L 磷酸二氫鉀溶液-三乙胺(100∶0.1),梯度洗脫(0~5min,A 相15%→15%;5~15 min,A 相 15%→75%;15~25 min,A 相 75→15% );檢測波長:262 nm;進樣量:20 μL。
2.2 溶液制備
對照品溶液:取馬來酸氯苯那敏對照品11.82 mg,精密稱定,置100 mL容量瓶中,加30%甲醇溶液溶解并稀釋至刻度,搖勻,作為對照品貯備液。精密量取5 mL,置25 mL容量瓶中,加30%甲醇溶液稀釋至刻度,搖勻,即得。
供試品溶液:取復方銀翹氨敏膠囊20粒,計算平均裝量,取適量(約相當于馬來酸氯苯那敏1.13 mg),精密稱定,置50 mL容量瓶中,加30%甲醇溶液20 mL,超聲處理10 min,加30%甲醇溶液稀釋至刻度,搖勻,濾過,取續濾液,作為供試品溶液(供含量測定用)。另取本品1粒,置50mL容量瓶中,加30%甲醇溶液20mL,超聲處理10 min,加30%甲醇溶液稀釋至刻度,搖勻,濾過,取續濾液,作為供試品溶液(供含量均勻度檢查用)。
陰性對照品溶液:按處方配制不含馬來酸氯苯那敏的陰性樣品,按供試品溶液制備方法配制,即得。
2.3 方法學考察
專屬性試驗:分別精密吸取馬來酸氯苯那敏對照品溶液、供試品溶液(含量測定用)和陰性對照品溶液各10 μL,照2.1項下色譜條件進樣,記錄色譜圖。詳見圖1。
線性關系考察:取馬來酸氯苯那敏對照品11.44 mg,精密稱定,置25 mL容量瓶中,加30%甲醇溶液溶解并稀釋至刻度,搖勻,制成質量濃度為 456.23 μg/mL 的對照品貯備液。精密量取上述對照品貯備液適量,以30%甲醇溶液為溶劑,分別制成質量濃度為4.56,18.25,45.62,68.43,91.24,114.06 μg /mL 的溶液,分別精密量取10 μL,注入液相色譜儀,按2.1項下色譜條件進樣測定,以對照品溶液質量濃度(X)為橫坐標、峰面積(以氯苯那敏計,下同,Y)為縱坐標進行線性回歸,得回歸方程 Y =13.933 X-0.506 2,r=0.999 9(n=6)。結果表明,馬來酸氯苯那敏質量濃度在4.56~114.06 μg/mL范圍內與峰面積線性關系良好。
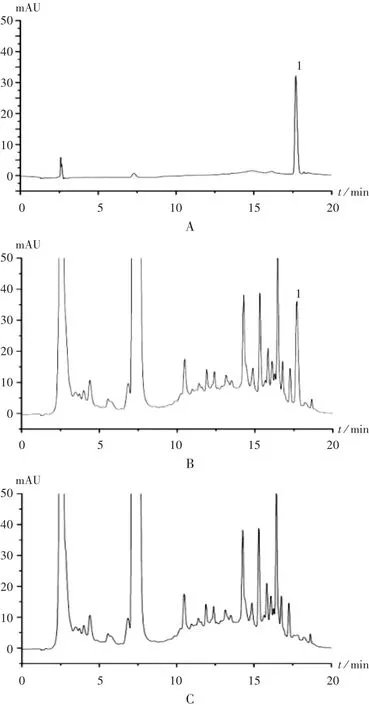
圖1 高效液相色譜圖
重復性試驗:取批號為170311的復方銀翹氨敏膠囊,照2.2項下方法平行制備6份供試品溶液,分別精密量取各溶液10 μL,每份進樣2次,記錄色譜圖。結果馬來酸氯苯那敏平均含量為標示量的102.34%,RSD為1.30%(n=6),表明方法重復性良好。
精密度試驗:精密量取前述質量濃度為45.62μg/mL的對照品溶液10 μL,注入液相色譜儀,重復進樣6次,記錄色譜圖。結果的 RSD為0.18%(n=6),表明儀器精密度良好。
穩定性試驗:取批號為170311的樣品,依法制備供試品溶液,室溫放置,分別于 0,2,4,6,8,12,24 h 時進樣,測定峰面積。結果的 RSD為0.20% (n=7),表明供試品溶液在24 h內穩定性良好。
加樣回收試驗:取批號為170311且已知含量的樣品適量(約相當于馬來酸氯苯那敏0.6 mg),精密稱定,分別置6個50 mL容量瓶中,分別精密加入線性關系考察項下對照品貯備液1 mL,依法制備供試品溶液,精密量取10 μL,注入液相色譜儀,記錄峰面積,外標法計算含量。結果見表1。
2.4 樣品測定
含量測定:取3種批號的復方銀翹氨敏膠囊各3 份,依法制備供試品溶液,精密量取 10 μL,照 2.1 項下色譜條件測定,記錄峰面積,計算標示含量。結果3批樣品中馬來酸氯苯那敏的平均標示含量分別為102.61%,104.20%,103.84%(n =3)。
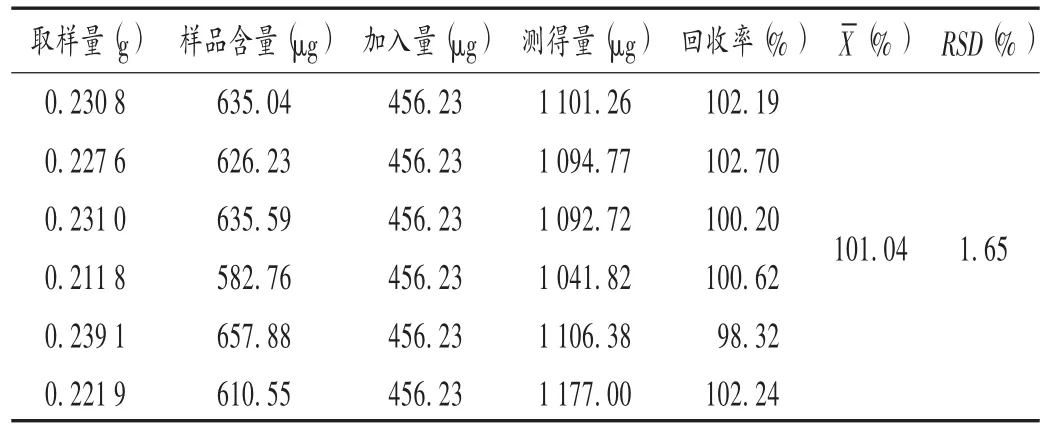
表1 馬來酸氯苯那敏加樣回收試驗結果(n=6)
含量均勻度測定:取3種批號的樣品各10粒,依法制備供試品溶液,精密量取各10 μL,照2.1項下色譜條件測定,記錄色譜峰峰面積(以氯苯那敏計),按外標法計算含量。結果見表2,符合2015年版《中國藥典(二部)》通則0941含量均勻度項下有關規定(A+2.2 S≤L,L = 15.0)。
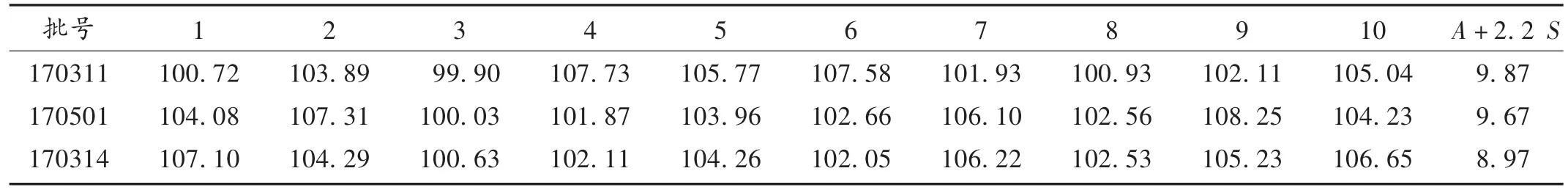
表2 樣品含量均勻度測定結果(n=3)
3 討論
3.1 檢測波長及流動相選擇
根據對照品溶液紫外光譜圖分析,馬來酸氯苯那敏在215 nm和262 nm波長處有最大吸收,同時參考2015年版《中國藥典(二部)》馬來酸氯苯那敏含量測定項下的色譜條件[8],最終選擇262 nm為檢測波長。比較流動相 B分別為 0.02 mol/L磷酸二氫鉀溶液或0.02 mol/L 磷酸二氫鉀溶液 -三乙胺(100 ∶0.1)對色譜峰的影響。結果當流動相B為后者時,氯苯那敏峰形有明顯提高,且基線平穩。
3.2 洗脫方式選擇
比較了等度洗脫和梯度洗脫的分離效能。當采用流動相A-流動相B(30∶70)等度洗脫時,氯苯那敏峰與其他相鄰成分峰不能達到有效分離,而在梯度洗脫程序下,氯苯那敏峰分離度好,且保留時間在20 min內,故采用梯度洗脫方法更適宜。
3.3 定量峰選擇
在研究中的色譜條件下,馬來酸氯苯那敏出現馬來酸與氯苯那敏2個峰[9-12],但馬來酸出峰時間早,易與溶劑峰和維生素C色譜峰重疊,且氯苯那敏為有效成分,故選擇氯苯那敏峰進行定量。
