稀土銪雙功能螯合劑的合成
2018-10-31 02:05:48趙啟含常宇馬玉芹潘利華
長春理工大學學報(自然科學版) 2018年5期
趙啟含,常宇,馬玉芹,潘利華
(1.長春理工大學 化學與環境工程學院,長春 130022;2.中國科學院長春應用化學研究所,長春 130022)
均相時間分辨熒光免疫分析(HTRF)是一種快速、簡便且抗干擾性強的分析技術,是將熒光共振能量轉移(FRET)技術和時間分辨熒光免疫分析技術(TRFIA)相結合而形成的一種新的方法[1-3]。TRFIA是以稀土離子螯合劑為標記物進行檢測分析,利用鑭系元素的熒光特性可以有效地排除非特異熒光的干擾。而HTRF是在其基礎上利用免疫反應生成免疫復合物的標記信號與未反應的標記物信號之間明顯的差異進行分析檢測的,所以在溶液中不需分離處理便可直接進行測定。HTRF具有操作簡單、高特異性、高靈敏度、高穩定性、水溶性等優異特點[4-7],在臨床醫學、化學分析等領域[8-11]受到大量關注。
HTRF中設計合成具有高穩定性、高靈敏度、高特異性、良好水溶性的穴狀雙功能稀土螯合劑是極具挑戰性的工作。該類螯合劑的雙功能性表現在可同時與蛋白質和稀土離子連接,連接后,不會影響蛋白質活性且具備稀土離子的優異性質。
本實驗從HTRF的需求考慮,設計制備一種稀土銪雙功能螯合劑。以2,6-二甲基吡啶-4-羧酸甲酯為原料,合成2,6-{N,N',N,N'-[二(2,2’-聯吡啶-6,6’-二甲基)]二氨甲基}-吡啶-4-羧酸-Eu3+,通過IR、MS等表征方法確認各合成產物的結構。并對其光譜性質進行研究。螯合劑的結構及合成路線如圖1所示。
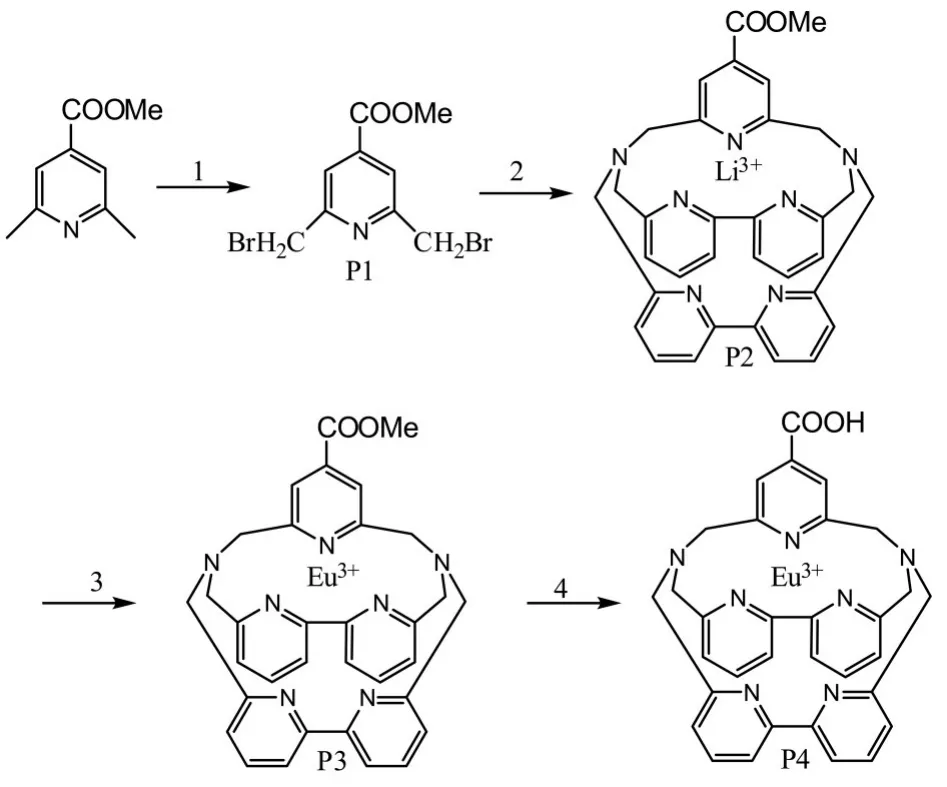
圖1 所合成的配體及螯合劑的結構及合成路線
1 實驗
1.1 主要儀器和試劑
FTSB5傅立葉紅外光譜儀(上海大中分析儀器廠);DSC-7差熱分析儀(美國Pericin Elmer公司);7.0 T型超導磁體型傅里葉變換離子回旋共振質譜儀(美國IonSpec公司);Unity-400MHz核磁共振儀(美國Varian公司);Hitachi F-7000型熒光光譜儀(日本HAMAMATSU公司);C10027型光子多通道分析儀(日本HAMAMATSU公司);FLSP-920型研究級光譜儀(英國愛丁堡儀器公司);熒光壽命測量儀(課題組自主研制)。
2,6-二甲基吡啶-4-羧酸甲酯(濟南某公司);N-溴代丁二酰亞胺(肯特催化有限公司);偶氮二異丁腈(江蘇全威化工);中性氧化鋁(國藥試劑)。C18反相硅膠(濟南博納生物技術)三氯甲烷、三氟乙酸、甲醇、叔丁醇等所用試劑規格均為AR級純度。
1.2 中間體合成
1.2.1 2,6-二溴甲基吡啶-4-羧酸甲酯(化合物P1)的合成
無水無氧條件下,將2,6-二甲基吡啶-4-羧酸甲酯(1g,5.54mmol),1.91g N-溴代丁二酰亞胺(NBS),四氯化碳(100ml)置于反應瓶中,攪拌10min,原料一部分溶解,加入0.04g偶氮二異丁腈(AIBN)。當溫度為40℃時,50W鹵素燈進行激發照射,將體系溫度升高至77℃,在回流狀態下反應2h,冷卻至室溫,用40ml飽和NaHCO3溶液洗,除去反應過程中產生的琥珀酰亞胺[12],三氯甲烷萃取(20ml×4),有機相用無水Na2SO4干燥。隔天過濾,并在真空狀態下除去溶劑,得到棕黃色油狀液體。以柱層析方式純化分離(淋洗劑:二氯甲烷),得到純凈的化合物P1。總收量215.4mg,總產率24.14%,封好放于干燥器內備用。
1.2.2 2,6-{N,N',N,N'-[二(2,2'-聯吡啶-6,6'-二甲基)]二氨甲基}-吡啶-4-羧酸甲酯(化合物P2)的合成
無水無氧的條件下,將N,N',N,N'-[二(2,2'-聯吡啶-6,6'-二甲基)]二胺(133mg,0.338mmol)溶于乙腈(267ml)中,攪拌10min(固體一部分溶解),向溶液中加入251mg的Li2CO3,提升溫度至反應液回流(80℃~83℃),60min后。向體系中滴加原料2化合物P1的乙腈溶液{P1(110mg,0.341mmol)+乙腈(67ml)},控制滴加速率,2h內滴完。繼續反應30h。冷卻反應液至30℃,用250ml沙漏抽濾,真空狀態下蒸干溶劑。用柱層析方式純化分離(淋洗劑為V二氯甲烷:V甲醇=9:1),得到90.4mg的純凈的化合物P2,產率46.05%,封好放于干燥器內備用。
1.3 螯合劑合成
1.3.1 2,6-{N,N',N,N'-[二(2,2'-聯吡啶-6,6'-二甲基)]二氨甲基}-吡啶-4-羧酸甲酯-Eu3+(化合物P3)的合成
氮氣保護的狀態下,將原料化合物P2(40mg,5.68×10-5mol)溶解于 10ml的無水甲醇中,固體全溶,攪拌狀態下,將預先制備的EuCl3.6H2O(23.6mg)加入到反應瓶中,溶液呈無色透明狀,升高溫度至溶液沸騰回流(65℃~68℃),23h后反應結束,冷水浴冷卻至室溫,加入5ml乙醚,出現白色的絮狀沉淀,抽濾、真空狀態下將沉淀物烘干,得到乳白色固體粉末32 mg,即化合物P3,不進行純化處理,封好放于干燥器內備用。
1.3.2 2,6-{N,N',N,N'-[二(2,2'-聯吡啶-6,6'-二甲基)]二氨甲基}-吡啶-4-羧酸-Eu3+(化合物P4)的合成
將原料化合物P3在堿性條件下水解,除去甲酯基團,取化合物P3(20mg)溶于3mL無水CH3OH中,室溫下攪拌,溶液呈無色透明狀,將1mol/L的LiOH的水溶液0.1mL滴入反應體系,溶液變白色渾濁,再加入0.5mL的1mol/L的LiOH水溶液,渾濁度增大,室溫下反應3.5h,蒸干反應溶劑,以C18反相硅膠為固定相進行柱層析純化,對(乙腈:1%三氟乙酸水溶液=7:3,V:V),得到乳黃色粉末固體9mg,即Eu3+-螯合劑(化合物P4),產率為43.44%,粉末在紫外燈下照射可發出明亮的橘紅色光,封好放于干燥器內備用。
1.3.3 化合物P4(螯合劑)溶液的配制
取0.81mg的化合物P4,用2ml的二次水溶解,固體全溶后,再用二次水定容到10ml的容量瓶中,配制成濃度為1×10-4mol/L的螯合劑水溶液。封好,用于檢測螯合劑的激發光譜、熒光光譜以及熒光壽命。
2 結果與討論
2.1 合成方法優化
2.1.1 化合物P1
利用NBS對原料溴化制備化合物P1,主要是NBS的溴自由基脫離并與原料進行連接的過程,此類反應過程中不僅需要光的照射激發還要用引發劑來促進反應的進行[8],本文選用AIBN作為引發劑,并研究了不同光源照射以及NBS使用量等對化合物P1產率的影響。
(1)不同光源照射對化合物P1產率的影響
確定其他反應條件不變,研究不同光源照射對反應后化合物P1產率的影響。
用100W照明燈與50W鹵素燈分別進行試驗,兩種燈光激發照射下,反應均可進行,但是照明燈在達到與鹵素燈相同產率時,所用時間較長(4h,鹵素燈為1h),兩種燈光照射均用薄層色譜法(TLC)檢測,可以發現鹵素燈照射反應進程很快。雖產生副產物點稍多,但對化合物P1產率幾乎無影響,這可能是因為鹵素燈照射促進了溴自由基的生成,在較短的時間內即可產生大量的溴自由基,加快了反應進程。因此,綜合考慮,本實驗選用50W鹵素燈進行照射。
(2)NBS使用量對化合物P1產率的影響
在其他反應條件為最佳的情況下,研究不同NBS使用量對反應后化合物P1產率的影響,研究結果如表1所示。

表1 NBS使用量對化合物1產率的影響
當NBS:2,6-二甲基吡啶-4-羧酸甲酯為1:1.9時化合物P1的產率最高。NBS含量過少的原因是,反應進行不完全,但是NBS過多時,反應液中溴自由基含量過多,生成過多的副產物(-CBr3/-CHBr2),使目標產物產率降低。
2.1.2 反應時間對化合物P2的影響
在其他反應條件為最佳的情況下,對化合物P2的反應時間進行優化,結果如表2所示。

表2 反應時間對化合物P2產率的影響
當反應時間為22h、26h時,反應不完全,目標產物產率較低,當反應時間為30h時,目標產物產率最高為46.05%。
2.2 合成產物的表征
2.2.1 2,6-二溴甲基吡啶-4-羧酸甲酯(化合物P1)的表征
圖2所示為2,6-二溴甲基吡啶-4-羧酸甲酯(化合物 P1)的質譜,MS(70eV)m/z(%):(M+H)+=324.0與化合物P1加氫的分子量理論值相符。
圖3所示2,6-二溴甲基吡啶-4-羧酸甲酯(P1)的核磁氫譜,δ:3.98有單峰,為酯中甲基的3個氫;δ:4.59有單峰為溴取代甲基中的4個氫,δ:7.92處的單峰為吡啶環中的2個氫,1H-NMR譜給出的各氫總數、出峰位置和化合物P1結構吻合。
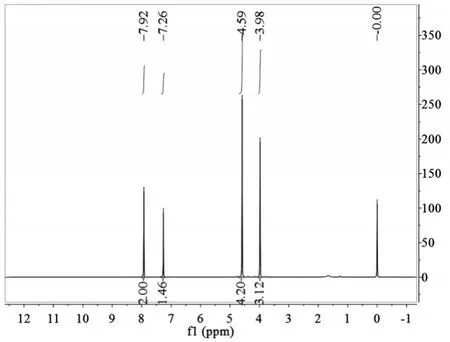
圖3 2,6-二溴甲基吡啶-4-羧酸甲酯的核磁(1H-NMR)譜圖
圖4所示為2,6-二溴甲基吡啶-4-羧酸甲酯(P1)的紅外光譜(a)與原料2,6-二甲基吡啶-4-羧酸甲酯的紅外光譜(b)對比可知,在616~582cm-1出現C-Br鍵的特征吸收峰,說明原結構中的-CH3被溴化成功,其他特征峰位置基本不變。
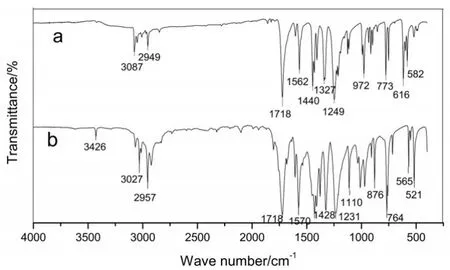
圖4 化合物P1(a)及其原料(b)的紅外光譜(IR)
2.2.2 2,6-{N,N ′,N,N ′-[二(2,2 ′-聯吡啶-6,6′-二甲基)]二氨甲基}-吡啶-4-羧酸甲酯(化合物P2)的表征
圖5所示為化合物P2的核磁氫譜1H-NMR(以CDCl3作為測試溶劑,射頻電磁波的頻率為400MHz),δ:3.95~4.10分別為酯中甲基的3個氫以及6個亞甲基中的12個氫共15個氫;δ:7.41~7.90為吡啶環中的14個氫,1H-NMR譜給出的氫個數、特征峰位置和其分子結構相符。化合物P2的分子量理論值是555,圖6所示為化合物P2的質譜圖,MS(70eV)m/z(%):(M+Na)+=578.2與化合物P2加鈉的分子量理論值相符。
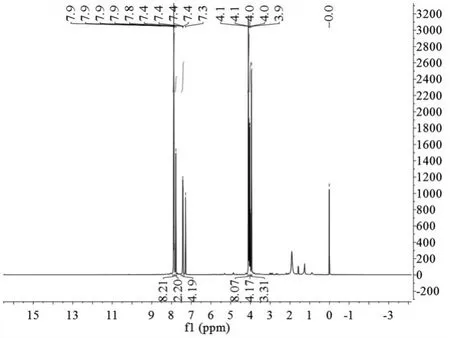
圖5 化合物P2的1H NMR譜
2.2.3 2,6-{N,N',N,N'-[二(2,2’-聯吡啶-6,6’-二甲基)]二氨甲基}-吡啶-4-羧酸甲酯-Eu3+(化合物P3)的表征
化合物P3的分子量理論值為708,圖7所示為化合物P3的質譜,MS(70eV)m/z(%):(L+Eu3++2Cl-)+=778.0與化合物P3加上稀土離子Eu3+以及中和其正電荷的兩個氯離子Cl-的分子量理論值相符。
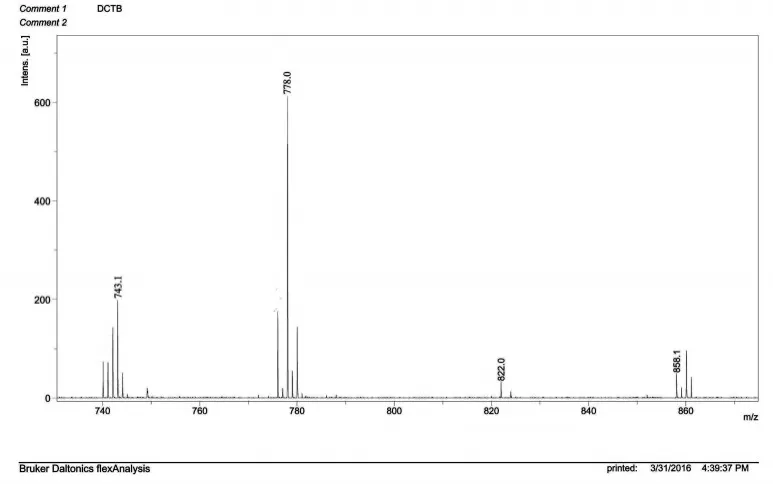
圖7 化合物P3的質譜(MS)
2.2.4 螯合劑2,6-{N,N',N,N'-[二(2,2'-聯吡啶-6,6'-二甲基)]二氨甲基}-吡啶-4-羧酸-Eu3+(化合物P4)的表征
螯合劑P4的分子量理論值是694.1,圖8所示為Eu3+-螯合劑(化合物P4)的質譜,MS(70eV)m/z(%):(L+Eu3+-H++CF3COO-)+=806.1與其加上CF3COO-的分子量理論值相符。
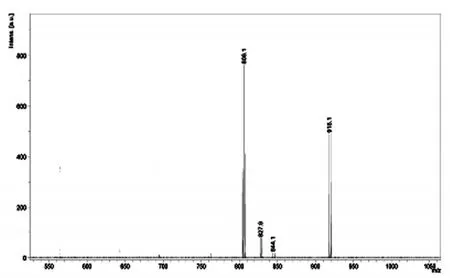
圖8 Eu3+-螯合劑(化合物P4)的質譜(MS)
2.3 Eu3+-螯合劑(化合物P4)的熒光光譜分析
取1×10-4mol/L的Eu3+-螯合劑(化合物P4)溶液液,稀釋其濃度至10-5mol/L,室溫下測試其熒光性質,測試數據如表3所示。
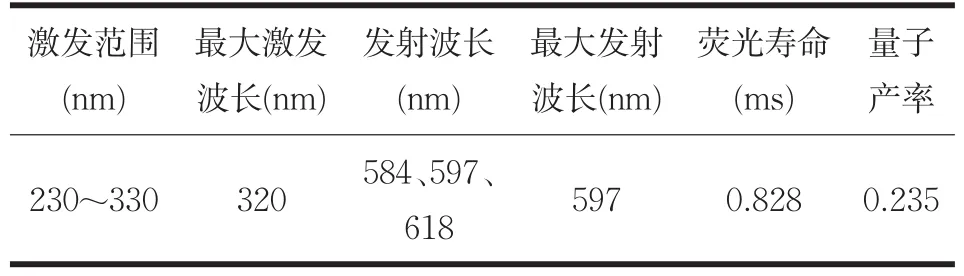
表3 化合物P4的熒光性質
圖9所示為Eu3+-螯合劑(化合物P4)在600V電壓下的激發光譜和熒光光譜,該螯合劑的激發范圍是230~330nm,最大激發波長為320nm,發射波長為 584nm(5D0-7F1)、597nm(5D0-7F2)、618nm(5D0-7F3),最大發射波長為597nm(5D0-7F2[13]),激發和發射光譜間沒有重疊,stokes位移可達到277 nm,此Eu3+-螯合劑的熒光發射強度較高,發射峰尖銳。符合HTRF技術的分析要求。
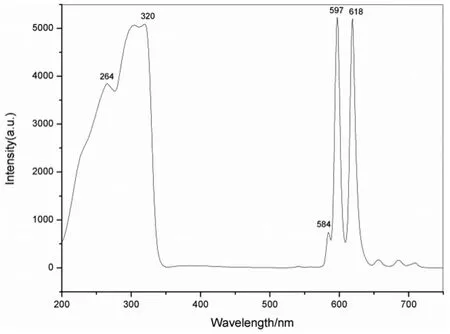
圖9 Eu3+-螯合劑的激發和熒光光譜
圖10所示為Eu3+-螯合劑(化合物P4)的熒光衰減曲線,通過單指數擬合可知此Eu3+-螯合劑的熒光壽命為0.828ms,可以有效地避免本底熒光的干擾,符合HTRF的分析要求。
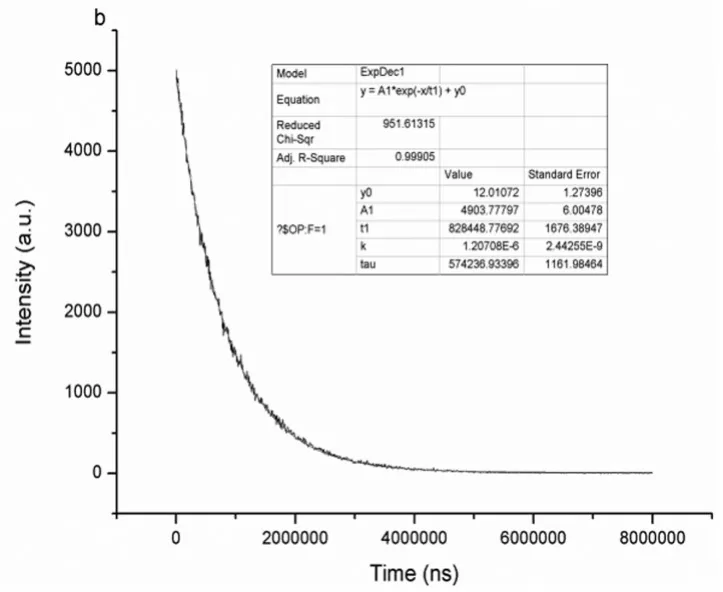
圖10 Eu3+-螯合劑的熒光衰減曲線
量子產率Φ是所有處于激發態的稀土螯合劑經過輻射躍遷返回基態的分子個數與其總分子數的比值,一般用量子產率表達熒光螯合物的熒光發射強度,其值越大,強度越強,分析技術中一般要求熒光標記物的量子產率在0.1~1之間。該Eu3+-螯合劑的熒光量子產率為0.235(322nm氙燈激發),可以達到分析技術中的應用要求,Eu3+-螯合劑具有一定實際應用價值。
圖11所示為Eu3+-螯合劑(化合物P4)濃度梯度曲線,最初配制的濃度為1.12×10-4mol/L的溶液,經稀釋分別配成濃度為2.23×10-6mol/L、1.24×10-6mol/L、5.93×10-7mol/L、1.79×10-8mol/L進行濃度梯度檢測,該螯合劑在水中的摩爾濃度與其相對熒光強度呈線性正相關,相關系數為0.995,而且即使在摩爾濃度很低時,仍具有很強的熒光強度。
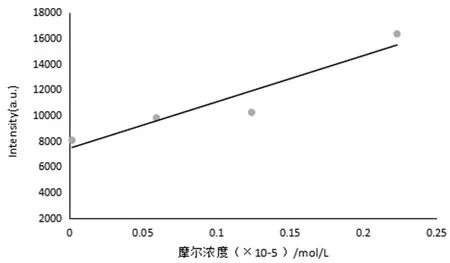
圖11 Eu3+-螯合劑的濃度梯度曲線
3 結論
本論文以2,6-二溴甲基吡啶-4-羧酸甲酯為原料經NBS溴化、酯水解等反應過程合成螯合劑2,6-{N,N',N,N'-[二(2,2’-聯吡啶-6,6’-二甲基)]二氨甲基}-吡啶-4-羧酸-Eu3+,并通過1H NMR、IR、MS、DSC等表征方法對各配體及螯合劑的結構進行了確認,并對螯合劑的光譜性質進行了研究。結果表明:該螯合劑的激發光譜譜帶較寬、最大激發波長為320nm,最大發射波長為597nm(5D0-7F2),激發光譜與熒光光譜間不重疊,熒光壽命高達828μs,熒光量子產率Φ為0.235。螯合劑在水中的摩爾濃度與其相對熒光強度呈正相關,相關系數為0.995,且在較低濃度下,該配體仍然具有很強的熒光強度,而且螯合劑具有較長的熒光壽命能夠有效的避免背景熒光的干擾,達到了時間分辨熒光的效果。綜上所述,該Eu3+-螯合劑可以滿足HTRF的分析要求。
