常染色體隱性遺傳性皮膚松弛癥的相關基因研究進展
2018-09-28 03:49:00石武娟薛珊珊
天津醫藥 2018年9期
關鍵詞:基因突變
石武娟,薛珊珊
皮膚松弛癥(cutis laxa,CL)是指因細胞外基質中的彈力蛋白合成受阻或結構異常引起的一種癥候群,是一種罕見的結締組織病[1]。該病的典型特征為非衰老所致的皮膚彈性下降、松弛及下垂,其中以面部皮膚受累最為多見,可伴有眼瞼、臉頰及頸部皮膚的松弛和下垂。同時,患者還可伴有其他富含彈性纖維的組織器官的功能障礙[2],如血管、心臟、肺等。因為皮膚松弛癥的遺傳方式和臨床表現具有多樣性,所以給患者及時準確的診治帶來一定的困難與挑戰。本文旨在綜述近年來常染色體隱性遺傳性皮膚松弛癥發病機制的研究進展,從而為該病的診治及預防提供新的思路。
1 皮膚松弛癥的分類
按遺傳特征,可將皮膚松弛癥分為遺傳性和獲得性兩大類。獲得性皮膚松弛癥是指某些因素如感染或藥物等作用使彈性蛋白結構和(或)數量發生不同程度的病理改變,最終導致皮膚彈性減退以致松弛[3-5]。獲得性皮膚松弛癥通常表現為局部病灶,且在發病前無皮膚損害的表現,但亦可出現全身受累[5]。與獲得性皮膚松弛癥相比,遺傳性皮膚松弛癥通常發病早,在出生時或嬰兒期即可發病。遺傳性皮膚松弛癥的發病率約為1/40萬~1/20萬[1],主要分為常染色體顯性遺傳、常染色體隱性遺傳及X連鎖隱性遺傳三種遺傳方式。
2 皮膚維持彈性的生物學基礎
皮膚的細胞外基質由多種成分共同構成,包括膠原、蛋白多糖、層粘連蛋白、原纖蛋白及彈性纖維等。其中,彈性纖維是最主要的成分,約占皮膚質量的2%~4%,為皮膚、肺、大血管等富含彈力纖維的器官提供彈性。彈性纖維主要由彈力蛋白核心與微纖維構成。微纖維則由微纖毛構成,其被覆于彈力蛋白核心的外周,為彈力蛋白的沉積提供支架。微纖毛包括衰老關鍵蛋白fibrillin和微纖毛相關的糖蛋白等[6]。fibullins主要分布于微纖維的內表面,主要參與彈力蛋白沉積到微纖毛支架上及微纖維與細胞表面相互作用的過程[6]。
彈力蛋白由成纖維細胞及平滑肌細胞以彈力蛋白原的形式合成和分泌。彈力蛋白原交聯過程在銅離子依賴的賴氨酰氧化酶(LOX)的介導下進行,其作為彈性纖維具備彈性及不溶性的基礎。大部分彈力蛋白是在胎兒發育過程中累積的,少部分在出生后積累[7-8]。
以上過程中的任何環節出現異常,都將影響其下游的信號通路,從而影響細胞與彈性纖維的相互作用,最終導致皮膚彈性下降。
3 常染色體隱性遺傳性皮膚松弛癥的分型及特點
常染色體隱性遺傳性皮膚松弛癥(autosomal recessive cutis laxa,ARCL)是發病率最高且臨床表現最為復雜的類型[9]。因其損害程度及受累器官變化多樣,加之臨床表現錯綜復雜,給臨床的診治造成巨大困難。筆者以下主要介紹ARCL最為常見的Ⅰ、Ⅱ、Ⅲ型的臨床特點及其相關的分子生物學研究進展。
3.1 ARCL-Ⅰ型ARCL-Ⅰ型的主要臨床表現包括典型重度肺氣腫和致死性血管病變,其最常見臨床表現包括肺不張、肺氣腫、胃腸道憩室、泌尿生殖系統憩室及血管發育異常。血管發育異常主要表現為動脈瘤、纖維肌性動脈發育不良和狹窄等[10-11]。此外,顱骨異常、囟門閉合延遲、關節松弛及髖關節脫臼、腹股溝疝等也是常見臨床表現[12-13]。學者們一致認可依據其致病基因,將其分為ⅠA(ARCL-ⅠA)、ⅠB(ARCL-ⅠB)及ⅠC(ARCL-ⅠC)三種[14]。研究發現,衰老關鍵蛋白4(FBLN4)和FBLN5基因突變常引起嚴重的肺氣腫,但不伴血管迂曲或動脈瘤的形成[12];而人潛伏轉化生長因子β結合蛋白4(LTBP4)基因突變患者的胃腸道及泌尿生殖道的臨床癥狀更嚴重。
3.1.1 ARCL-ⅠAARCL-ⅠA是由Fibulin-5(FBLN5)突變導致的皮膚松弛癥。Fibulin-5是Fibulin家族成員之一,Fibulin家族成員廣泛分布于富含彈力蛋白的組織中,能直接與彈力蛋白結合,或者連接原纖蛋白的微纖維,從而在彈力纖維及細胞外基質的形成過程中發揮重要作用。其編碼基因定位于人染色體的14q32.1,所編碼蛋白是一種含有448個氨基酸的糖蛋白,分子質量約為60 ku。Fibulin-5作為細胞外基質蛋白,主要分布于大血管、心臟瓣膜、主動脈弓、肺、子宮及皮膚等含豐富彈性纖維的器官,因此,當Fibulin-5基因突變時,其他富含Fibulin-5的器官也會受累,從而表現出相應的臨床癥狀。
3.1.1.1 臨床表現ARCL-ⅠA主要臨床特征為嚴重的發育性肺氣腫,常常表現為致死性的新生兒呼吸窘迫,其他伴隨癥狀包括主動脈瓣膜狹窄,肺動脈狹窄及腹股溝疝等。
3.1.1.2 分子生物學機制 研究發現Fibulin-5具有多種生物學功能,包括調控類賴氨酰氧化酶-1(LOXL1)、LOXL2、LOXL4、原纖蛋白-1、彈力蛋白原、LTBP2及LTBP4等分子的生物學功能,見圖1[15]。以上分子與Fibullin-5結合不僅可促進彈力蛋白原凝聚及交聯,同時有助于形成彈性纖維。此外,Fibulin-5突變后亦可影響蛋白的折疊及分泌功能,進而導致彈力蛋白不能與微纖毛整合。因此,當Fibullin-5基因突變致其功能受損時,Fibullin-5參與的上述過程均受到影響,最終導致皮膚松弛癥的發生,見圖2。
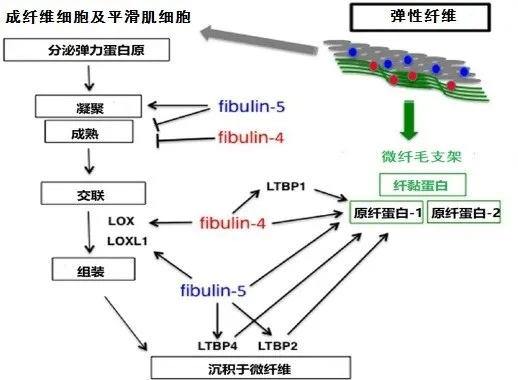
Fig.1 The pathogenesis genes involved in ARCL-typeⅠregulated the formation of elastic fibers圖1 ARCL-Ⅰ型的各致病基因調控彈力纖維形成過程示意圖
3.1.2 ARCL-ⅠBARCL-ⅠB的致病基因為Fibulin-4(FBLN4)。Fibulin-4與Fibulin-5屬于同一家族成員,其編碼基因定位于人染色體的11q13.1-q13.2區域,所編碼蛋白是一種由442個氨基酸組成的細胞外基質蛋白,分子質量約50 ku。Fibulin-4不僅參與彈力纖維的形成過程,而且對于膠原的微纖毛形成過程具有重要的調控作用[12,16]。Fibulin-4兩個等位基因部分或全部突變,均會因Fibulin-4蛋白水平的顯著降低或缺如而引起嚴重的臨床癥狀,大部分患者在出生后18個月內夭折,這一結論與體外實驗研究結果一致[17]。Fibulin-4突變患者的臨床癥狀依Fibulin-4蛋白功能受影響的程度而呈現多樣性。
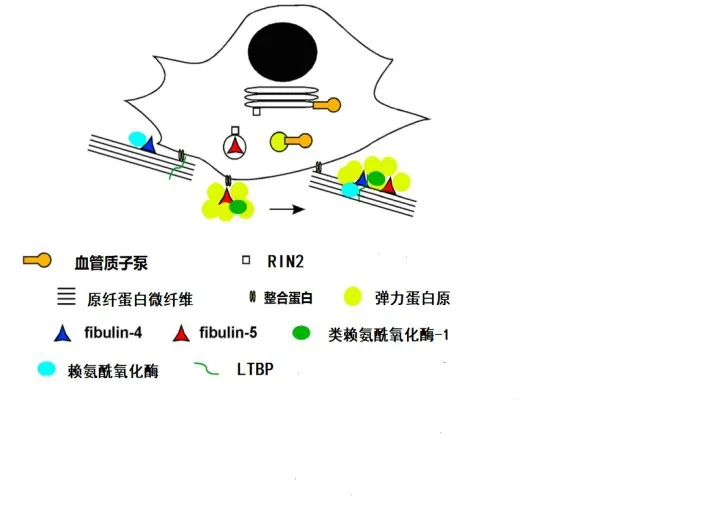
Fig.2 The molecular biological mechanism of regulating elastic fibers involved the ARCL-typeⅠ圖2 ARCL-Ⅰ型的相關致病分子調控彈力纖維形成的分子生物學機制
3.1.2.1 臨床表現FBLN4突變所致的ARCL-ⅠB可累及全身各系統,主要臨床表現包括動脈迂曲、動脈瘤、肺動脈高壓、發育性肺氣腫、脆骨病、關節松弛、膈疝及腹股溝疝等[10,13]。
3.1.2.2 分子機制Fibulin-4可與彈力蛋白原、原纖蛋白-1及LTBP1相結合[18],通過影響這些分子,進而影響彈力纖維的形成。Fibulin-4與Fibulin-5的功能區分主要通過選擇與不同的配體結合發揮各自調節作用。若兩者均與同一種分子結合,則主要通過結合細胞類型的差異或親和力的不同來確保兩者在彈力纖維的組裝過程中各司其職,互不替代(具體過程見圖1)。例如,Fibulin-5對于彈力蛋白原的親和力要高于原纖蛋白-1,而Fibulin-4則剛好相反,對原纖蛋白-1的親和力要高于彈力蛋白原。Fibulin-4與彈力蛋白原、LOX形成三聚體,易化彈力蛋白的交聯作用[19-20];而Fibulin-5則加強彈力蛋白原的凝聚作用[21]。Fibulin-4和Fibulin-5均調控彈性蛋白原凝聚成熟過程中的顆粒大小[21]。Fibulin-5缺乏可導致彈力蛋白的顆粒直徑過大,且不能與微纖維支架整合[15,22](具體過程見圖2)。與此相反,Fibulin-4突變則導致彈性纖維數量顯著減少,表現為彈力蛋白的交聯過程受阻及彈力纖維的結構紊亂[20],最終導致相應的臨床癥狀(具體過程見圖2)。
3.1.3 ARCL-ⅠCARCL-ⅠC也被稱為Urban-Rifkin-Davis綜合征,由LTBP4突變造成。LTBP4在3種不同啟動子作用下產生3種亞型,1個小的LTBP4S及2個大的LTBP4L。在人類發現LTBP4突變時其三種亞型均受到影響,而在小鼠只發現LTBP4S異常。LTBP4S完全敲除小鼠的彈性纖維表型與人類完全缺乏的表現一致,比如發育性肺氣腫及彈力纖維形態異常,即彈力蛋白表面光滑而無微纖毛[23]。但LTBP4突變造成的胃腸道及泌尿生殖道的癥狀目前只在人類觀察到,在LTBP4S完全敲除的小鼠并未發現,這一現象提示消化系統及泌尿生殖道的發育可能主要依賴于LTBP4L[24]。
3.1.3.1 臨床表現 嚴重的發育性肺氣腫、憩室病、胃腸道的擴大和(或)狹窄、膀胱憩室、膈疝及腹股溝疝均是其常見的臨床表現[25]。LTBP4突變的患者常死于呼吸衰竭及腸穿孔。大量研究證實LTBP4具備兩種功能,一是調控轉化生長因子(TGF)-β及其下游信號通路的作用,一是有助于彈性纖維的組裝[26]。
3.1.3.2 分子機制LTBP4不同亞型的功能具有差異性,如LTBP4L偏向于與TGF-β結合,從而調控其下游信號通路;而LTBP4S則傾向于與細胞外基質的形成密切相關[27]。LTBP4可與Fibulin-5、原纖蛋白-1結合,從而有助于Fibulin-5與彈力蛋白形成的復合體錨定到微纖毛,見圖1。LTBP4也被報道與纖黏蛋白及硫酸乙酰素蛋白多糖(HSPGs)相互結合,在細胞黏附過程中發揮著至關重要的作用[28]。人LTBP4編碼C端細胞黏附區域的基因突變導致微纖毛束變厚和彎曲,提示LTBP4在調控細胞與細胞外基質分子沉積到彈力纖維的過程中具有至關重要的作用[25]。
3.2 ARCL-Ⅱ型
3.2.1 臨床表現 該型的主要表現為廣泛性皮膚松弛、皺褶,但面部癥狀通常較輕。特征性臨床癥狀為前囟閉合延遲、前額突出、輕度尖顱、V型眉、眼裂下斜、齲齒、智力缺陷,常伴有宮內發育遲緩、臀部錯位、雞胸、脊柱側凸等骨骼異常、腹股溝疝和扁平足[29-30]。依據其突變分為ⅡA(ATP6V0A2基因突變)和ⅡB(PYCR1基因突變)兩型。
3.2.2 ARCL-ⅡAARCL-ⅡA由編碼囊泡內質子泵V-ATP酶亞基的ATP6VOA2基因突變導致。ATP6VOA2基因突變導致N-連接和O-連接的糖基化異常,囊泡轉運受阻,最終導致彈力蛋白原積聚于高爾基體,呈現多系統代謝性疾病。目前臨床上可通過監測血漿中的轉鐵蛋白或載脂蛋白CⅢ的N-連接糖基化及O-連接糖基化水平,進而評估ATP6VOA2突變程度[31]。
3.2.2.1 臨床表現ARCL-ⅡA的主要臨床表現包括嚴重的神經系統缺陷,如巨腦回、小頭畸形、肌無力、癲癇、近視、神經退行性病變及Dandy-Walker畸形。臨床特征為全身廣泛的皮膚松弛、囟門閉合延遲或不閉合、尖顱、關節過伸,同時具備神經系統異常癥狀及面部畸形的表現。典型的神經系統異常癥狀包括發育延遲、智力障礙、肌無力、小頭畸形、失聰、癲癇,甚至頭磁共振提示腦實質呈鵝卵石樣發育不良。典型的面部畸形特征表現為前額突出、V型眉、眼裂下斜、長人中、面頰下垂及朝天鼻。此外,也可出現胎兒宮內發育遲緩、藍色鞏膜、漏斗胸、腹股溝疝、扁平足及先天性髖關節脫位等癥狀,部分患者亦可患有近視或斜視等眼部疾患。
3.2.2.2 分子機制 臨床檢測發現N-連接和O-連接的糖基化蛋白水平異常,即:含4個葉酸殘基的轉鐵蛋白水平減少,而含2個或3個葉酸的轉鐵蛋白水平顯著增加,以上生化指標的異常提示N-連接糖基化的蛋白水平顯著增加。此外,包含2個葉酸殘基的血漿載脂蛋白CⅢ減少,含單個葉酸殘基的轉鐵蛋白含量增加,這兩種現象均提示ATP6VOA2基因突變[32]。新生兒在圍生期轉鐵蛋白水平可能處于正常范圍內,但隨著日齡增加,轉鐵蛋白異常的表現愈發明顯。由此可見,生化檢測是區分ARCL-ⅡA與ARCL-ⅡB的重要方法,對于ARCL-Ⅱ型的分型具有重要的指導意義。
3.2.3 ARCL-ⅡBARCL-ⅡB由線粒體酶PYCR1基因突變造成。該型主要由參與脯氨酸代謝的PYCR1基因突變導致線粒體功能異常[33]。PYCR1編碼的酶主要功能是以NAD(P)H-依賴的方式將5-羧酸二氫吡咯轉變為脯氨酸[34],從而影響線粒體的能量代謝過程。該型常見的臨床表現為腹部及背部的皮膚松弛、先天性髖關節脫位、胎兒宮內或生后生長發育遲緩、骨質疏松、三角形面容或早衰面容、蒜頭鼻、下頜突出、內眥贅皮、藍色鞏膜,大耳及小頭畸形等。神經系統表現為肌無力、發育遲緩、智力障礙、胼胝體發育不全或缺如,極少數可能智力正常。
3.3 ARCL-Ⅲ型ARCL-Ⅲ型,又稱為De Barsy綜合征,該型主要由乙醛脫氫酶18家族成員A1(ALDH18A1)基因突變造成[35-36]。ALDH18A1編碼的蛋白主要功能是將谷氨酸轉變為脯氨酸、鳥氨酸及精氨酸的生物合成過程中的中間代謝產物5-半醛谷氨酸。若ALDH18A1基因突變,則脯氨酸、鳥氨酸及精氨酸三種氨基酸的生物合成過程都將受到影響,因此,多系統都將受累并表現出相應的臨床癥狀。該型主要以頭發稀少、角膜異常、胎兒宮內發育遲緩和皮膚松弛等早衰外觀為特征。患者出生時表現為皮膚薄、半透明、多皺褶和無彈性并伴有明顯的可見靜脈。眼睛特征性變化為角膜鮑氏膜彈性纖維降解導致角膜混濁[36]。大部分可同時伴有神經及骨骼系統的異常,包括認知和(或)語言功能受損、髖關節脫位、關節過伸、脊柱側彎和足部畸形。部分患者與Ⅱ型有重疊的特征,但肌張力障礙和角膜異常提示為本型。
筆者總結了遺傳性皮膚松弛癥的各分型及其臨床特征、致病基因及功能,見表1。皮膚松弛癥的預后及干預措施因其遺傳類型的不同而差異顯著。常染色體顯性遺傳性皮膚松弛癥(autosomal dominant cutis laxa,ADCL)和X連鎖遺傳性皮膚松弛癥(X-linked cutis laxa,XLCL)根據患者的臨床表現及其家族史較易鑒別診斷。ARCL因常伴嚴重的心血管系統損害,患兒多于兒童期死亡。與ADCL和XLCL相比,ARCL更依賴于有效的產前指導及咨詢以降低其發病率。隨著醫學研究的發展及轉化醫學的廣泛應用,可通過檢測某些疾病的致病基因或相關蛋白以指導疾病的早期診斷及治療,如目前通過生化檢測轉鐵蛋白水平區分ARCL-ⅡA與ARCL-ⅡB等。因此,探究與疾病相關的基因及信號通路來指導ARCL的防治及早期診斷值得進一步研究探討。
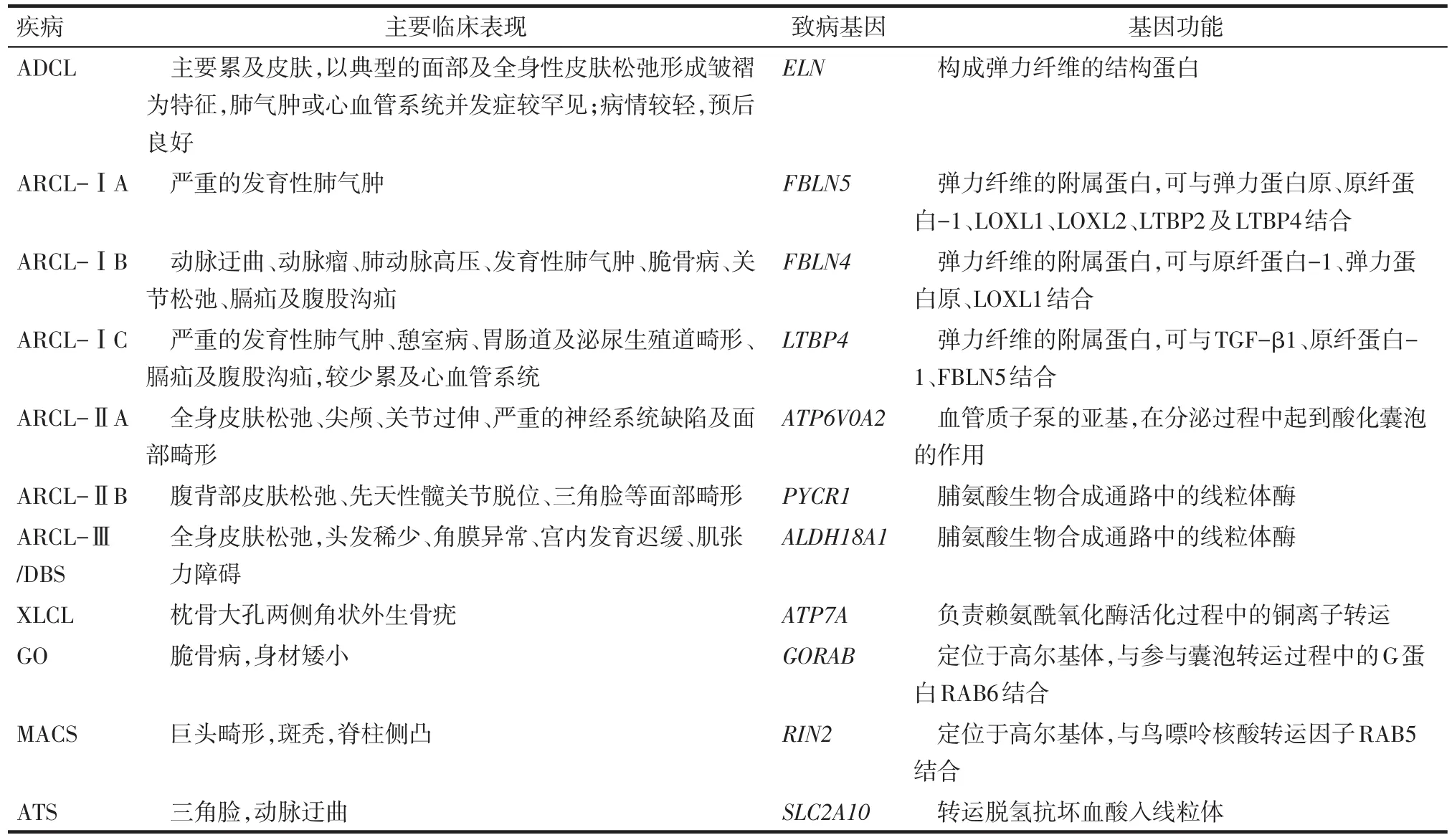
Tab.1 The specific manifestations of common types of cutis laxa and the corresponding genes and functions表1 常見皮膚松弛癥的類型及其特征性的臨床表現、相應的致病基因及其功能
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
