H3K27me3聚集MEG3 lncRNA啟動(dòng)子通過MDM2/p53途徑誘導(dǎo)多發(fā)性骨髓瘤RPMI8226細(xì)胞凋亡逃逸
2018-07-04 10:32:20董娟娟彭萬仁顏兵雪錢婷婷蔡謎謎孫國(guó)平
安徽醫(yī)科大學(xué)學(xué)報(bào) 2018年7期
關(guān)鍵詞:水平
董娟娟,彭萬仁,顏兵雪,錢婷婷,蔡謎謎,王 華, 孫國(guó)平
多發(fā)性骨髓瘤(multiple myeloma, MM)是造血干細(xì)胞的惡性克隆性疾病,具有極強(qiáng)的異質(zhì)性,涉及諸多發(fā)病環(huán)節(jié)。MM發(fā)生涉及細(xì)胞增殖、凋亡、分化、周期轉(zhuǎn)換等異常過程中,組蛋白甲基化/去甲基化修飾平衡功能紊亂發(fā)揮舉足輕重的作用[1-5]。組蛋白甲基化修飾狀態(tài)與細(xì)胞凋亡的發(fā)生關(guān)系密切。研究[6]顯示,長(zhǎng)鏈非編碼RNA(long noncoding RNA, lncRNA)作為新型表觀遺傳學(xué)調(diào)控方式,可與組蛋白甲基化相互調(diào)節(jié)。通過此方式兩者共同參與染色質(zhì)重構(gòu)、轉(zhuǎn)錄激活、轉(zhuǎn)錄干擾等多種重要的調(diào)控過程。母系表達(dá)基因3 (materially expressed gene 3,MEG3) lncRNA是一個(gè)被證實(shí)具有腫瘤抑制功能的長(zhǎng)鏈非編碼RNA,在57%的MM患者中表達(dá)缺失[7],但其作用機(jī)制尚不清楚。現(xiàn)將重點(diǎn)探討MM中MEG3 lncRNA基因啟動(dòng)子區(qū)域組蛋白H3賴氨酸殘端27位的三甲基化(H3K27me3)表達(dá)情況,明確基因啟動(dòng)子區(qū)域H3K27me3修飾紊亂是否導(dǎo)致MM中MEG3 lncRNA轉(zhuǎn)錄缺失,從而為MM的臨床治療尋找新的方向。
1 材料與方法
1.1藥品與試劑MTT、碘化丙啶(PI)、DMSO、RNaseA、Hoechst 33258熒光染液均購自美國(guó)Sigma公司產(chǎn)品; RNA抽提試劑TRIzol、RPMI 1640培養(yǎng)基均購自美國(guó)Gibco公司; 胎牛血清(FBS)購自杭州四季青生物工程材料有限公司; Annexin-V FITC/PI 試劑盒購自南京凱基生物科技發(fā)展有限公司; 引物由上海生物工程技術(shù)服務(wù)有限公司合成。逆轉(zhuǎn)錄-聚合酶鏈反應(yīng)(RT-PCR)試劑盒購自日本Toyobo 公司; 抗體Zeste基因增強(qiáng)子同源物2(enhancer of Zeste homolog 2,EZH2)、H3K27me3、鼠雙微體基因2(murine double minute 2,MDM2)、p53和ubiquitin-p53購自美國(guó)Cell Signaling公司; 氯仿、乙醇、異戊醇為國(guó)產(chǎn)分析醇,購自杭州長(zhǎng)征化學(xué)試劑有限公司。
1.2儀器BD流式細(xì)胞儀、Bio-Rad M450 酶聯(lián)免疫檢測(cè)儀購自美國(guó)CA公司; ChemiDocXRS 蛋白條帶密度分析軟件購自美國(guó)Bio-Rad 公司;Real-time PCR儀器購自羅氏公司。
1.3細(xì)胞培養(yǎng)RPMI8226細(xì)胞購自武漢大學(xué)保藏中心,用含100 U/ml鏈霉素、100 U/ml青霉素、10%滅活FCS的RPMI1640培養(yǎng)基, 在5% CO2、37℃、飽和濕度條件下培養(yǎng), 根據(jù)細(xì)胞數(shù)量,每2~3 d 換液傳代1次, 倍增時(shí)間24~48 h, 取細(xì)活性>95%的對(duì)數(shù)生長(zhǎng)期細(xì)胞用于后續(xù)各項(xiàng)實(shí)驗(yàn)。
1.4MTT比色法檢測(cè)細(xì)胞增殖收集處于對(duì)數(shù)生長(zhǎng)期的RPMI8226細(xì)胞,調(diào)整細(xì)胞濃度至2×105/ml,在96孔板中每孔加入體積200 μl該細(xì)胞懸液,邊緣孔用無菌PBS填充。細(xì)胞分為MDM2拮抗劑干預(yù)組、p53拮抗劑干預(yù)組和DMSO干預(yù)組, 按要求時(shí)間在5% CO2、37 ℃、飽和濕度條件下孵育,每孔加入 5 mg/ml 的 MTT 20 μl, 繼續(xù)37 ℃孵育4~6 h。離心棄上清液,小心用PBS沖2~3次后,每孔加入 150 μl DMSO, 置搖床上低速振蕩10 min,在藍(lán)色結(jié)晶充分溶解后, 在Bio-Rad M450酶聯(lián)免疫檢測(cè)儀以O(shè)D492 nm波長(zhǎng)測(cè)量各孔的吸光度(A)值, 重復(fù)實(shí)驗(yàn) 3次, 按公式:抑制率=1-實(shí)驗(yàn)組A值/對(duì)照組A值,計(jì)算細(xì)胞增殖抑制率。
1.5Annexin-VFITC/PI雙標(biāo)法流式細(xì)胞術(shù)檢測(cè)細(xì)胞凋亡按實(shí)驗(yàn)設(shè)計(jì)收集經(jīng)siRNA-EZH2處理后RPMI8226細(xì)胞,同時(shí)設(shè)計(jì)陰性對(duì)照組(DMSO處理), 經(jīng)4 ℃預(yù)冷的PBS離心洗2次, 按1×106/ml密度于100 μl 的 buffer結(jié)合緩沖液中重懸細(xì)胞, 分別加入 Annexin-V FITC 5 μl和PI 10 μl , 震蕩混勻, 室溫避光條件下反應(yīng)15 min, 再加入buffer結(jié)合緩沖液至300 μl, 樣品制備完畢后上流式細(xì)胞儀檢測(cè)。
1.6RT-PCR法檢測(cè)EZH2、MEG3lncRNA及MDM2和p53表達(dá)按試劑盒說明書合成cDNA。按以下條件進(jìn)行逆轉(zhuǎn)錄反應(yīng):30 ℃、10 min, 42 ℃、20 min, 99 ℃、5 min, 4 ℃、5 min, -20 ℃保存。EZH2引物序列:上游引物5′-GGACCACAGTGTTACCAAGCAT-3′,下游引物5′-GTGGGGTCTTTATCCGCTCAG-3′, 擴(kuò)增產(chǎn)物為79 bp;MDM2 引物序列:上游引物5′-CAGTAGCAGTGAATCTACAGGGA-3′, 下游引物5′-CTGATCCAACCAATCACCTGAAT-3′, 擴(kuò)增產(chǎn)物為79 bp;MEG3lncRNA引物序列:上游引物5′-ATCATCCGTCCACCTCCTTGTCTTC-3′,下游引物5′-GTATGAGCATAGCAAAGGTCAGGGC-3′, 擴(kuò)增產(chǎn)物為284 bp; β-actin 引物序列:上游引物5′-TGAGACCTTCAACACCCCAG-3′,下游引物5′-GCCATCTCTTGCTCGAAGTC-3′, 擴(kuò)增產(chǎn)物為206 bp;以cDNA為模板, 進(jìn)行MEG3 lncRNA、MDM2的PCR擴(kuò)增。取擴(kuò)增產(chǎn)物5 μl, 經(jīng)瓊脂糖凝膠電泳、紫外線照相、Bandscan軟件進(jìn)行掃描分析, 得出目的基因的灰度值進(jìn)行半定量分析。
1.7Westernblot檢測(cè)EZH2、H3K27me3、MDM2及p53蛋白表達(dá)將各處理組細(xì)胞收集至50 μl加入苯甲基磺酰氟和蛋白酶抑制劑的冷細(xì)胞裂解液中,以提取細(xì)胞總蛋白。按照Bio-Rad 試劑盒使用說明書標(biāo)定Marker蛋白及各泳道,電泳。在300 mA電流15 min的條件下轉(zhuǎn)膜,取出硝酸纖維素膜置于5%脫脂牛奶中4 ℃過夜封閉。加入稀釋的抗靶蛋白抗體與膜共同孵育(37 ℃)1 h。取出纖維素膜洗滌后,再與辣根過氧化物酶標(biāo)記的二抗共孵育(37 ℃)1 h。洗滌后,曝光,X光片掃描定影,輸入計(jì)算機(jī)。
1.8ChIP-RealtimePCR技術(shù)收集各處理組細(xì)胞;細(xì)胞固定,直接將甲醛加入細(xì)胞培養(yǎng)液,搖晃固定10 min后,迅速加入2.5 mol/L甘氨酸解交聯(lián)5 min,PBS洗滌,刮下細(xì)胞。超聲打碎:使用超聲水浴鍋,EP管在冰水混合物中超聲,將長(zhǎng)鏈的DNA打碎成200~1 000 bp的DNA片段。4 ℃、5 000 r/min離心5 min后去上清液。免疫沉淀:加入ChIP級(jí)H3K27me3抗體和對(duì)照IgG,和結(jié)合抗體的磁珠(或protein A/G)。經(jīng)稀釋后,超聲產(chǎn)物分成兩份,分別加入ChIP級(jí)H3K27me3抗體和對(duì)照IgG,輪轉(zhuǎn)過夜,第2天加入磁珠輪轉(zhuǎn)4 h。洗滌:順序分別用稀釋緩沖液、低鹽溶液、高鹽溶液、氯化鋰溶液以及TE緩沖液洗滌,共兩個(gè)循環(huán)。用洗脫緩沖液在65 ℃洗脫,過夜。DNA純化:將洗脫的產(chǎn)物用DNA純化試劑盒純化,得到50 μl純化產(chǎn)物DNA。
根據(jù)MEG3 lncRNA基因區(qū)域上游2 000~3 000 bp范圍分段設(shè)計(jì)引物,每300 bp設(shè)計(jì)一套,共合成8~10套引物。將ChIP以后的Input、IgG、IP三個(gè)樣本分別用10個(gè)引物進(jìn)行半定量PCR的篩選,如果IP組的樣本某套引物能P出條帶,說明H3K27me3和MEG3 lncRNA啟動(dòng)子區(qū)域的這段序列存在結(jié)合。再運(yùn)用QPCR定量富集倍數(shù):將半定量電泳PCR篩選出來的有結(jié)合的引物,再用Input和IP樣本進(jìn)行QPCR驗(yàn)證,計(jì)算得出ChIP富集的倍數(shù)。
1.9siRNA干擾技術(shù)轉(zhuǎn)染前1 d,常規(guī)消化細(xì)胞,制成細(xì)胞懸液、計(jì)數(shù),在6孔板內(nèi)鋪上1.2×105個(gè)細(xì)胞,加入1 500 μl 無抗生素培養(yǎng)基培養(yǎng) 24 h,到轉(zhuǎn)染時(shí)使細(xì)胞達(dá)到 30%~50%的匯合率。三組分別將EZH2-siRNA(EZH2為H3K27me3特異性的組蛋白甲基轉(zhuǎn)移酶)、siRNA 陰性對(duì)照、NC siRNA各5 μl溶于250 μl無血清的 Opti-MEM溶液中,輕柔混合均勻。將5 μl Lipofectamine RNA IMAX溶于250 μl無血清的Opti-MEM中,混合均勻,在室溫下孵育5 min后,將上述兩種混合物輕輕混合均勻,并在室溫下孵育20 min。注意混合試劑時(shí),請(qǐng)勿劇烈吹打或振蕩,手指輕彈管壁即可,用力過度可能會(huì)破壞siRNA的結(jié)構(gòu)。在6孔板中棄去舊培養(yǎng)基,每孔加入1 500 μl 的無血清培養(yǎng)基,并將上述兩種混合物加入6孔板,上下左右輕輕晃動(dòng)板子使得復(fù)合物混合均勻。細(xì)胞在37 ℃、5% CO2培養(yǎng)箱中培養(yǎng),在加完復(fù)合物4~6 h后,可更換完全培養(yǎng)基。48 h后,收集細(xì)胞,提取蛋白,測(cè)量靶蛋白EZH2及H3K27me3的表達(dá)水平或進(jìn)行其他實(shí)驗(yàn)。

圖1 RPMI8226細(xì)胞中下調(diào)H3K27me3的表達(dá)可以誘導(dǎo)RPMI8226細(xì)胞凋亡
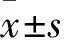
2 結(jié)果
2.1H3K27me3導(dǎo)致MM細(xì)胞凋亡逃逸EZH2是特異性的H3K27me3組蛋白甲基化轉(zhuǎn)移酶,合成特異性的EZH2-siRNA, 運(yùn)用電轉(zhuǎn)染技術(shù)使EZH2-siRNA進(jìn)入RPMI8226細(xì)胞內(nèi)(同時(shí)設(shè)有陰性對(duì)照)特異性抑制EZH2的表達(dá),運(yùn)用Western blot技術(shù)檢測(cè)干擾后EZH2、H3K27me3的表達(dá),結(jié)果顯示,通過干擾EZH2的表達(dá)可以明顯抑制H3K27me3的表達(dá),見圖1A。 Annexin-V FITC/PI雙標(biāo)法檢測(cè)H3K27me3對(duì)RPMI8226細(xì)胞凋亡的影響:早期凋亡細(xì)胞顯示為Annexin-V FITC標(biāo)記為陽性,同時(shí)PI標(biāo)記為陰性的細(xì)胞;然而,晚期凋亡細(xì)胞則為Annexin-V FITC與PI標(biāo)記均為陽性的細(xì)胞群。與正常RPMI8226細(xì)胞組對(duì)比,EZH2-siRNA干預(yù)組H3K27me3蛋白表達(dá)明顯降低,見圖1A,凋亡的RPMI8226細(xì)胞數(shù)目明顯增多。正常RPMI8226細(xì)胞組的早期凋亡率、晚期凋亡率分別為(0.13±0.02)%和(0.26±0.02)%;EZH2-siRNA干預(yù)組的早期凋亡率、晚期凋亡率分別為(1.39±0.27)%和(19.9±0.71)%;正常組和干預(yù)組間細(xì)胞凋亡率有明顯差異(t=4.870,P<0.05),見圖1B。
2.2ChIPReal-timePCR證實(shí)H3K27me3直接結(jié)合于MEG3lncRNA基因啟動(dòng)子區(qū)本研究以RPMI8226細(xì)胞為樣本進(jìn)行ChIP實(shí)驗(yàn)。結(jié)果顯示,在上千個(gè)基因啟動(dòng)子區(qū)域有高密度聚集的H3K27me3,其中包含很多癌相關(guān)基因。同時(shí)發(fā)現(xiàn)MEG3lncRNA啟動(dòng)子區(qū)存在H3K27me3聚集。進(jìn)一步運(yùn)用ChIP Real-time PCR證實(shí)MEG3lncRNA啟動(dòng)子區(qū)存在H3K27me3聚集,使用Graphpad Prism 5.0 軟件進(jìn)行統(tǒng)計(jì)分析,H3K27me3組較Control IgG組明顯差異,提示H3K27me3可直接結(jié)合于MEG3lncRNA基因啟動(dòng)子區(qū),見圖2。

圖2 H3K27me3可直接結(jié)合于MEG3lncRNA基因啟動(dòng)子區(qū)
2.3通過調(diào)控H3K27me3表達(dá)可上調(diào)RPMI8226細(xì)胞MEG3lncRNA表達(dá)水平RPMI8226細(xì)胞經(jīng)EZH2-siRNA干預(yù)組作用后,細(xì)胞內(nèi)MEG3lncRNA表達(dá)水平呈明顯升高,Control組 與siRNA-NC組干預(yù)后相比,兩組間MEG3lncRNA表達(dá)水平無明顯變化。但EZH2-siRNA干預(yù)后MEG3lncRNA表達(dá)水平是前者2倍,與Control組比較差異有統(tǒng)計(jì)學(xué)意義(P<0.05),見圖3。

圖3 抑制H3K27me3可上調(diào)RPMI8226細(xì)胞MEG3lncRNA表達(dá)水平
2.4通過調(diào)控RPMI8226細(xì)胞MEG3lncRNA表達(dá)可影響MDM2蛋白、mRNA水平RPMI8226細(xì)胞經(jīng)MEG3lncRNA-siRNA干預(yù)組作用后,細(xì)胞內(nèi)MDM2 mRNA表達(dá)水平呈明顯升高,Control組 與siRNA-NC組干預(yù)后相比,兩組間MDM2 mRNA表達(dá)水平無明顯變化。但MEG3lncRNA-siRNA干預(yù)后MDM2 mRNA表達(dá)水平是前兩者2倍,差異有統(tǒng)計(jì)學(xué)意義(P<0.05)。MEG3lncRNA-siRNA明顯上調(diào)mRNA表達(dá)水平的同時(shí),同時(shí)有效誘導(dǎo)了MDM2蛋白表達(dá)。MEG3lncRNA-siRNA干預(yù)組作用后,細(xì)胞內(nèi)MDM2蛋白水平呈明顯升高趨勢(shì),差異有統(tǒng)計(jì)學(xué)意義(t=3.269,P<0.05)。siRNA-NC組干預(yù)后MDM2蛋白水平無明顯變化,見圖4。
2.5RPMI8226細(xì)胞中可通過拮抗MDM2相關(guān)的p53泛素化導(dǎo)致p53降解以MDM2拮抗劑(Nutlin-3)干預(yù)RPMI8226細(xì)胞后,應(yīng)用Western blot檢測(cè)MDM2拮抗劑(Nutlin-3)對(duì)RPMI8226細(xì)胞的Ub-p53及p53蛋白水平調(diào)節(jié)變化;結(jié)果顯示,Nutlin-3可在蛋白水平上抑制p53的泛素化,故Ub-p53表達(dá)水平降低,使得p53降解被抑制,p53蛋白表達(dá)水平明顯升高,見圖5。Nutlin-3處理組與RPMI8226細(xì)胞未處理組(Control組)相比,差異有統(tǒng)計(jì)學(xué)意義(t=93.59,P<0.05)。

圖4 RPMI8226細(xì)胞經(jīng)MEG3lncRNA-siRNA干預(yù)后MDM2 mRNA和蛋白表達(dá)水平均明顯升高
A:各組Western blot中MDM2蛋白水平;B:各組PCR中MDM2基因水平;與Control組比較:*P<0.05

圖5 MDM2拮抗劑(Nutlin-3)干預(yù)后RPMI8226細(xì)胞的p53與Up-p53表達(dá)
3 討論
表觀遺傳學(xué)在腫瘤發(fā)生、發(fā)展中作用堪比傳統(tǒng)遺傳學(xué),表觀遺傳修飾的異常是所有人類腫瘤的共同特點(diǎn)。組蛋白甲基化酶和組蛋白去甲基化酶是表觀遺傳學(xué)的重要內(nèi)容,機(jī)體通過兩者共同作用動(dòng)態(tài)調(diào)節(jié)組蛋白甲基化模式,組蛋白甲基化模式是發(fā)揮表觀遺傳作用的重要方式之一,嚴(yán)格調(diào)控細(xì)胞的分化、增殖,維持機(jī)體細(xì)胞生命狀態(tài)的穩(wěn)定, 在細(xì)胞凋亡、周期等生命過程中發(fā)揮重要作用[8], 其失平衡則導(dǎo)致腫瘤的發(fā)生。H3K27me3 生物體細(xì)胞中廣泛富集于基因啟動(dòng)子區(qū)域, 介導(dǎo)染色質(zhì)重構(gòu),染色質(zhì)閉鎖,基因轉(zhuǎn)錄停止。也有報(bào)道稱除基因啟動(dòng)子區(qū)域外, H3K27me3尚可存在于結(jié)構(gòu)基因多個(gè)位點(diǎn),維持染色質(zhì)的濃縮狀態(tài)。
組蛋白甲基化酶復(fù)合物PRC2 的亞基 EZH2 在骨髓瘤中高表達(dá), 其調(diào)控的H3K27me3水平在腫瘤的發(fā)生發(fā)展中起重要作用,靶向抑制EZH2可增加瘤細(xì)胞的凋亡率。圖1A實(shí)驗(yàn)證實(shí),通過siRNA技術(shù)抑制MM RPMI8226細(xì)胞中EZH2表達(dá)后, H3K27me3表達(dá)量顯著下降,同時(shí)RPMI8226細(xì)胞凋亡率明顯增加。徐莉 等[9]也證實(shí)RPMI8226及H929細(xì)胞系中的H3K27me3高表達(dá)會(huì)導(dǎo)致本細(xì)胞凋亡相關(guān)分子caspase3、PARP均呈低表達(dá), 瘤細(xì)胞死亡率降低, 且同時(shí)對(duì)35例骨髓瘤患者標(biāo)本進(jìn)行Western blot分析, 證實(shí)EZH2、H3K27ME3的表達(dá)水平和骨髓瘤患者的疾病的惡性程度呈正相關(guān)。因此,推論H3K27me3的高表達(dá)保護(hù)了RPMI8226細(xì)胞的凋亡逃逸。
MEG3lncRNA作為第一個(gè)被發(fā)現(xiàn)有腫瘤抑制功能的lncRNA,證實(shí)在57% MM患者中表達(dá)缺失[7],但其作用機(jī)制尚不清楚。LncRNA是新近發(fā)現(xiàn)的具有強(qiáng)大調(diào)控功能的基因組“暗物質(zhì)”, 是一類轉(zhuǎn)錄長(zhǎng)度超過200 nt的RNA分子, 起初因無蛋白質(zhì)編碼功能,被認(rèn)為是基因組轉(zhuǎn)錄的“噪音”[10]。由于近年研究的不斷深入,lncRNA功能學(xué)發(fā)現(xiàn)突飛猛進(jìn), 已證實(shí)lncRNA參與了染色質(zhì)修飾、X染色體沉默、轉(zhuǎn)錄干擾、轉(zhuǎn)錄激活等多種重要的基因表達(dá)調(diào)控過程, 是重要的表觀遺傳學(xué)調(diào)控方式[11]。研究顯示lncRNA可與其他表觀遺傳形式相互作用,組蛋白甲基化可直接作用于lncRNA基因啟動(dòng)子區(qū)來調(diào)控lncRNA的表達(dá),反之,上游非編碼區(qū)轉(zhuǎn)錄出的lncRNA也可通過影響組蛋白甲基化/乙酰化等修飾狀態(tài)影響下游基因的表達(dá)。其表達(dá)缺失已被證實(shí)存在于多種腫瘤細(xì)胞系及實(shí)體腫瘤中。Benetatos et al[12]報(bào)道, 57%的MM病例MEG3 lncRNA基因啟動(dòng)子存在異常的甲基化譜,且伴隨MEG3 lncRNA表達(dá)缺失,并且缺失程度與MM的亞型和疾病的階段具有顯著相關(guān)性。然而, MEG3 lncRNA表達(dá)缺失的原因及其在MM的發(fā)生、發(fā)展中作用機(jī)制目前仍未有研究。本實(shí)驗(yàn)證實(shí)MM中H3K27me3可結(jié)合于MEG3 lncRNA基因啟動(dòng)子區(qū)域,且通過siRNA技術(shù)抑制H3K27me3甲基化酶EZH2降低H3K27me3表達(dá)的同時(shí),MEG3 lncRNA表達(dá)可明顯升高。因此推論,MM RPMI8226細(xì)胞中MEG3 lncRNA基因啟動(dòng)子區(qū)H3K27me3的異常高表達(dá),是MEG3 lncRNA表達(dá)缺失的重要原因。H3K27me3如何保護(hù)RPMI8226細(xì)胞的凋亡逃逸,是否與調(diào)控MEG3 lncRNA表達(dá)相關(guān)仍有待探索。
在神經(jīng)膠質(zhì)瘤中證實(shí),人為使MEG3 lncRNA過表達(dá)可抑制腫瘤細(xì)胞的DNA合成及細(xì)胞集落的形成,抑制MDM2表達(dá),誘導(dǎo)p53聚集,刺激p53介導(dǎo)的轉(zhuǎn)錄活化, 選擇性的調(diào)節(jié)p53下游靶基因 GDR-15(growth differentiation factor 15, GDF-15, 此基因具有抗增殖及腫瘤抑制功能)等的表達(dá), 從而抑制細(xì)胞增殖,誘導(dǎo)凋亡[13-17]。多發(fā)性骨髓瘤中,MEG3 lncRNA異常低表達(dá)是否同樣引起MDM2的異常活化及p53降解導(dǎo)致功能缺失,最終誘導(dǎo)MM逃避凋亡。本實(shí)驗(yàn)中證實(shí),RPMI8226細(xì)胞中H3K27me3表達(dá)被抑制時(shí),MEG3 lncRNA表達(dá)升高,同時(shí)MDM2在mRNA及蛋白水平上均明顯降低。同樣運(yùn)用siRNA技術(shù)證實(shí),MEG3 lncRNA表達(dá)被抑制時(shí),MDM2在mRNA及蛋白水平上均有所升高。本實(shí)驗(yàn)還證實(shí)運(yùn)用MDM2拮抗劑Nutlin-3可顯著降低蛋白水平泛素化p53的表達(dá),非泛素化p53的蛋白表達(dá)有升高趨勢(shì)。由此推論,MM中MEG3 lncRNA表達(dá)缺失解除了對(duì)MDM2表達(dá)的抑制,MDM2再通過與p53直接結(jié)合介導(dǎo)p53泛素化,間接促使p53的降解, 或直接抑制 p53 基因活化,導(dǎo)致 MM 細(xì)胞凋亡逃逸。
[1] Liu Y, Zeng L L, Chen Y,et al.Triptolide inhibits cell growth and induces G0-G1 arrest by regulating P21wap1/cip1 and P27kip1 in human multiple myeloma RPMI-8226 cells[J]. Chin J Cancer Res,2010,22(2):141-7.
[2] Zeng R,Zeng L,Chen Y,et al.Triptolide-induced apoptosis by inactivating nuclear Factor-kappa B apoptotic pathway in multiple myelomainvitro[J].J Huazhong Univ Sci Technolog Med Sci, 2011,31(4):446-51.
[3] Zhao F, Chen Y, Zeng L L,et al.Effects of triptolide on RIZ1 expression, proliferation, and apoptosis in multiple myeloma U266cells[J].Acta Pharmacol Sin,2010,31(6):733-40.
[4] Zhao F, Chen Y, Zeng L,et al.Role of triptolide in cell proliferation, cell cycle arrest, apoptosis and histone methylation in multiple myeloma U266 cells[J].Eur J Pharmacol,2010,646(1-3):1-11.
[5] Zhao F, Chen Y, Li R,et al.Triptolide alters histone H3K9 and H3K27 methylation state and induces G0/G1 arrest and caspase-dependent apoptosis in multiple myelomainvitro[J]. Toxicology,2010,267(1-3):70-9.
[6] Peffers M J, Balaskas P, Smagul A.Osteoarthritis year in review 2017:genetics and epigenetics[J].Osteoarthritis Cartilage,2017,26(3):304-11.
[7] Benetatos L, Dasoula A, Hatzimichael E,et al.Promoter hypermethylation of the MEG3 (DLK1/MEG3) imprinted gene in multiple myeloma[J].Clin Lymphoma Myeloma,2008,8(3): 171-5.
[8] Chen H,Landen C N, Li Y,et al.Epigallocatechin gallate and sulforaphane combination treatment induce apoptosis in paclitaxel-resistant ovarian cancer cells through hTERT and Bcl-2 down-regulation.[J].Exp Cell Res,2013,319(5):697-706.
[9] 徐 莉,陳協(xié)群.多發(fā)性骨髓瘤中組蛋白甲基化的分析研究[G].武漢:全國(guó)淋巴腫瘤診治進(jìn)展研討會(huì),2014.
[10] Bedoyareina O C, Ponting C P. Functional RNA classes: a matter of time[J].Nat Struct Mol Biol,2017,24(1):7-8.
[11] Bassett A R, Azzam G, Wheatley L,et al.Understanding functional miRNA-target interactionsinvivoby site-specific genome engineering[J].Nat Commun,2014,5:4640.
[12] Benetatos L,Dasoula A,Hatzimichael E,et al.Promoter hypermethylation of the MEG3 (DLK1/MEG3) imprinted gene in multiple myeloma[J].Clin Lymphoma Myeloma,2008,8(3):171-5.
[13] Wang P,Ren Z,Sun P.Overexpression of the long non-coding RNA MEG3 impairsinvitroglioma cell proliferation[J].J Cell Biochem,2012,113(6):1868-74.
[14] Zhou Y,Zhong Y,Wang Y,et al.Activation of p53 by MEG3 Non-coding RNA[J].J Biol Chem,2007,282(34):24731-42.
[15] Yan H,Yuan J,Gao L,et al.Long noncoding RNA MEG3 activation of p53 mediates ischemic neuronal death in stroke[J].Neuroscience,2016,337:191-9.
[16] Zhu J,Liu S,Ye F,et al.Long Noncoding RNA MEG3 interacts with p53 protein and regulates partial p53 target genes in hepatoma cells[J].Plos One,2015,10(10):e139790.
[17] Zhan R,Xu K,Pan J,et al.Long noncoding RNA MEG3 mediated angiogenesis after cerebral infarction through regulating p53/NOX4 axis[J].Biochem Biophys Res Commun,2017,490(3):700-6.
猜你喜歡
美與時(shí)代·美術(shù)學(xué)刊(2022年3期)2022-04-27 01:18:15
火花(2019年12期)2019-12-26 01:00:28
人大建設(shè)(2019年6期)2019-10-08 08:55:48
人大建設(shè)(2019年12期)2019-05-21 02:55:32
雜文月刊(2018年21期)2019-01-05 05:55:28
人大建設(shè)(2017年6期)2017-09-26 11:50:44
學(xué)苑創(chuàng)造·A版(2015年11期)2016-01-14 09:03:27
俄羅斯問題研究(2012年1期)2012-03-25 09:54:45
中國(guó)火炬(2010年12期)2010-07-25 13:26:22
中國(guó)火炬(2010年8期)2010-07-25 11:34:30
