空腹胰島素水平對高尿酸血癥病人痛風發作的預測價值
2018-06-19 06:16:54
精準醫學雜志 2018年3期
(青島大學附屬醫院內分泌與代謝性疾病科,山東 青島 266003)
痛風是由于血液中尿酸水平升高所致。尿酸水平較高時導致尿酸鹽結晶析出,晶體會沉積在關節、肌腱和周圍組織中[1]。目前認為,胰島素敏感組織(肌肉、脂肪組織和肝臟等)對胰島素的不敏感會加重慢性、低水平炎癥。炎癥被觸發后可以削弱胰島素的作用,胰島素的作用被削弱出現胰島素抵抗又會增強炎癥的反應性,兩者互相作用和影響,形成了一種惡性循環。炎癥反應與胰島素作用之間關系復雜。臨床實踐中發現許多合并高尿酸血癥和肥胖的糖尿病病人,在胰島素治療后,容易發生痛風,提示胰島素升高與痛風發作的誘發可能存在一定的關系。本研究旨在探討胰島素水平與痛風發作之間的關系,以及其可能誘發痛風的切點,以進一步了解痛風的發病機制,指導在治療合并高尿酸血癥或肥胖的糖尿病病人過程中如何適宜地使用胰島素。現將結果報告如下。
1 資料和方法
1.1 一般資料
選擇2016年6月—2017年8月在我院內分泌科因高尿酸血癥痛風發作入院,尚未進行降尿酸或緩解疼痛治療的病人78例(痛風組),均為男性,年齡為17~64歲,平均(41.29±13.33)歲。病人均符合2015年ACR/EULAR痛風分類標準。排除標準:骨髓增生性疾病、腫瘤放化療后、腎臟病等所導致的繼發性痛風病人。同時選擇高尿酸血癥無痛風并發癥的門診病人120例(高尿酸組)作為對照,均為男性,年齡17~84歲,平均(44.36±14.41)歲。兩組病人均無糖尿病、嚴重感染及肝腎疾病,且近期未服用利尿藥或影響胰島素及尿酸代謝的藥物,無大量飲酒史;兩組年齡、身高等比較,差異無顯著性(P>0.05)。
1.2 檢測指標及方法
入院后病人禁飲食8 h后于次日的清晨采集靜脈血,采用全自動生化分析儀(BECKMAN COULTER,AU5800,USA)檢測血清胰島素、尿酸、糖化血紅蛋白、谷丙轉氨酶/谷草轉氨酶(ALT/AST)、三酰甘油、總膽固醇、高密度脂蛋白(HDL)、低密度脂蛋白(LDL)及游離脂肪酸的水平。
1.3 統計學處理
2 結 果
2.1 兩組病人胰島素等指標比較
痛風組病人空腹胰島素水平、體質量指數、腰臀比與高尿酸組病人比較明顯升高,HDL水平顯著降低(t=-2.287~3.276,P<0.05);而兩組病人血尿酸、糖化血紅蛋白、ALT/AST、三酰甘油、總膽固醇、LDL、游離脂肪酸水平無明顯統計學差異(P>0.05)。結果見表1。
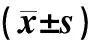
表1 兩組病人相關指標的比較
2.2 痛風發作相關危險因素分析
以是否發生痛風為自變量,以空腹胰島素、體質量指數、腰臀比、HDL為協變量,將樣本隨機抽取90%作為訓練樣本,利用二元Logistic回歸分析進行篩選得到兩個變量與痛風發作有統計學意義。空腹胰島素OR值大于1,表示空腹高胰島素水平是痛風發作的危險因素;而HDLOR值小于1,表示HDL水平增高是痛風發作的保護因素。其模型參數估計見表2。
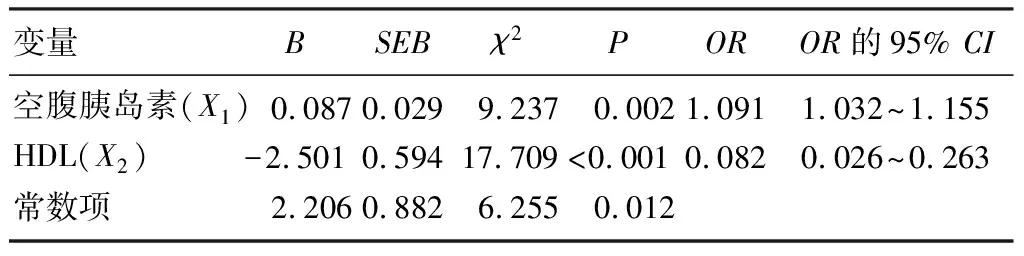
表2 Logistic回歸分析結果
進而可以建立預測模型:
根據此模型可以預測痛風發作的概率Pr,然后用ROC曲線決定判斷痛風發作的臨界點;取臨界點Pr為0.542,即當Pr≥0.542時判為痛風發作,當Pr<0.542時判為痛風不會發作;將剩余10%的樣本作為測試樣本回帶,對預測模型進行檢驗,得出預測模型診斷痛風發作的靈敏度為54.5%,特異度為87.5%。
2.3 空腹胰島素和HDL預測痛風的切點
以ROC曲線下面積0.5作為參考界值,空腹胰島素的ROC曲線下面積為0.80(圖1)。提示空腹胰島素可用于預測痛風發作。進一步分析顯示,以空腹胰島素10.618 mU/L為最佳臨界值,預測痛風發作的診斷靈敏度為80.0%,診斷特異度為65.5%,約登指數為0.455。HDL的ROC曲線下的面積則為0.381(圖2),提示HDL不適用于預測痛風發作。
3 討 論
本研究探討了空腹胰島素水平與痛風發作的關系。結果顯示,痛風病人組的空腹胰島素水平高于高尿酸組病人。研究表明,除了某些罕見單因素綜合征外,多種危險因素直接或間接作用引起痛風,主要包括高尿酸血癥、遺傳學因素及藥物等[2]。
腎小管功能受高胰島素血癥干擾,胰島素介導的葡萄糖清除下降使血清尿酸清除減低,從而導致高尿酸血癥[3]。血清胰島素升高可增加腎臟近端小管對尿酸的重吸收[4];高胰島素血癥可致細胞氧化磷酸化功能受損,腺苷的濃度升高可促進尿酸的生成并增加腎臟儲存尿酸的能力[5]。高水平胰島素及胰島素前體可促進腎小管Na+-H+交換,使尿酸重吸收相應增多而出現高尿酸血癥[6]。
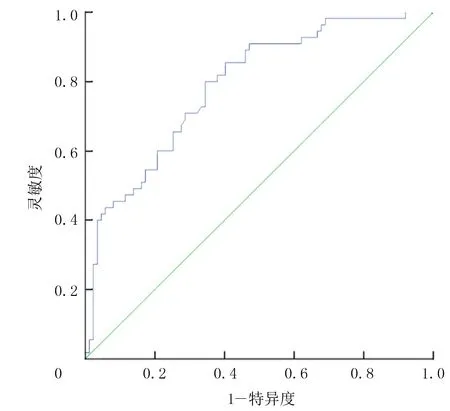
圖1 空腹胰島素的ROC曲線
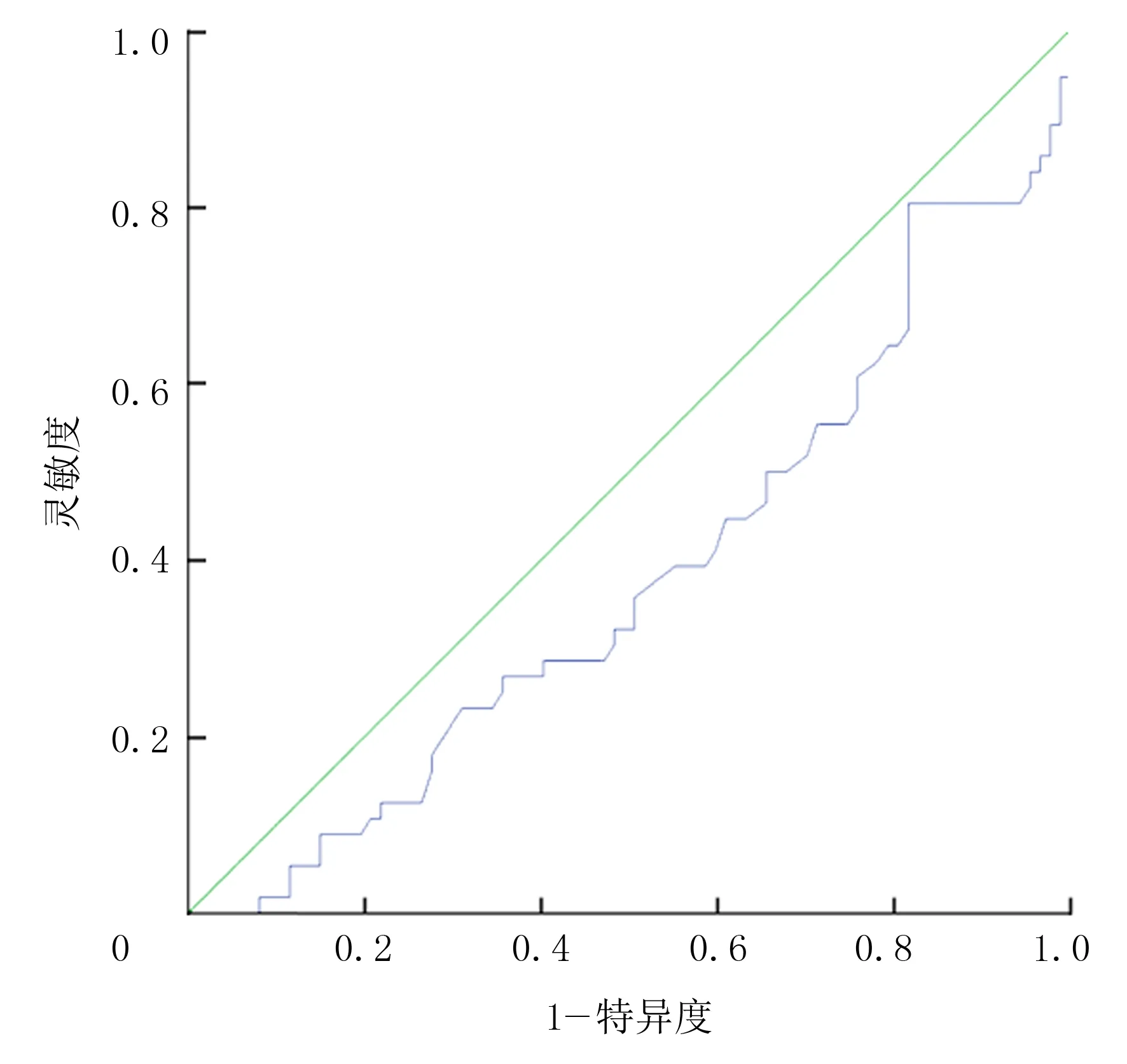
圖2 HDL的ROC曲線
血清胰島素水平升高可以激活PI3K,通過Akt/IKK信號通路激活參與非代謝器官炎癥反應的核心轉錄因子(NF-κB)[7]。活化的 NF-κB 進入細胞核后,參與細胞炎癥的過程,調節包括 TNF-α、IL-1β、IL-6、IL-8在內的炎癥因子的表達[8-9],而IL-1β在痛風性關節炎發生及痛風發作的過程中發揮關鍵的作用[10-13]。PI3K又可以通過AKt激活mTOR,進而調節如TNF-α、IL-1β等炎癥因子的分泌[14-16]。同時促炎因子又可激活JNK和IKK/NF-κB信號通路上調炎癥遞質水平,炎性遞質水平升高又反過來激活JNK和IKK/NF-κB,兩者相互影響,最終導致痛風發作[17]。高胰島素血癥還可以通過MAPK通路激活NF-κB加重炎癥的反應[7],導致痛風的發作。本研究通過Logistic回歸發現空腹高胰島素水平是痛風發作的危險因素,可能是通過上述機制導致痛風的發生。本研究計算的胰島素的ROC曲線下面積為0.800,提示空腹胰島素可用于預測痛風的發作。
高胰島素血癥通過增加ChREBP、SREBP-1c、低度炎癥反應以及TNF-α等促進肝臟合成脂質,脂質增多使新陳代謝中的核酸總量增加,加速嘌呤從頭合成途徑,增加尿酸生成[18]。脂肪分解為酮體的過程可阻礙尿酸排泄,使尿酸水平間接升高。關節腔內尿酸濃度過飽和形成尿酸鹽結晶,從而啟動痛風炎性通路,引發炎癥級聯反應[19]。脂質在脂肪細胞中過多的蓄積引發肥胖,從而導致脂肪組織的炎癥反應[20]。激活NF-κB使脂肪細胞增加趨化因子如MCP1的合成和分泌,這導致巨噬細胞的浸潤。高胰島素血癥還可以通過MAPK通路激活NF-κB加重炎癥的反應[7],從而導致痛風的發作。這與本研究得出的結論一致,可能是胰島素升高導致痛風發作的機制。
本研究還發現,HDL水平增高是痛風發作的保護因素。在促炎細胞因子介導的炎癥反應中HDL也起負性調節作用。HDL水平降低,其負性調節炎癥的作用也降低,可能在炎癥反應中加重痛風相關的損害。
總之,本研究結果顯示痛風組的空腹胰島素水平顯著高于高尿酸組,胰島素水平是痛風發作的危險因素,胰島素水平的升高預示著風險增加。HDL水平升高是痛風發作的保護因素。本研究進一步評估空腹胰島素預測痛風發作的切點。結果提示,空腹胰島素水平大于10.618 mU/L時,預測痛風發作的診斷靈敏度為80.0%,診斷特異度為65.5%。綜上所述,空腹胰島素水平有助于臨床預測病人痛風發作,為預防痛風發作提供一個很好指標。
[參考文獻]
[1] RICHETTE P, BARDIN T. Gout[J]. Lancet, 2010,375(9711):318-328.
[2] KUO C F, GRAINGE M J, ZHANG W, et al. Global epidemiology of gout: Prevalence, incidence and risk factors[J]. Nat Rev Rheumatol, 2015,11(11):649-662.
[3] NISKANEN L, LAAKSONEN D E, LINDSTROM J, et al. Serum uric acid as a harbinger of metabolic outcome in subjects with impaired glucose tolerance: The Finnish Diabetes Prevention Study[J]. Diabetes Care, 2006,29(3):709-711.
[4] DOSHI M, TAKIUE Y, SAITO H, et al. The increased protein level of URAT1 was observed in obesity/metabolic syndrome model mice[J]. Nucleosides Nucleotides Nucleic Acids, 2011,30(12):1290-1294.
[5] SUZUKI S, YONEYAMA Y, SAWA R, et al. Relation between serum uric acid and plasma adenosine levels in women with preeclampsia[J]. Gynecol Obstet Invest, 2001,51(3):169-172.
[6] TSUNODA S, KAMIDE K, MINAMI J, et al. Decreases in serum uric acid by amelioration of insulin resistance in overweight hypertensive patients: effect of a low-energy diet and an insulin-sensitizing agent[J]. Am J Hypertens, 2002,15(8):697-701.
[7] PARK M H, KIM D H, LEE E K, et al. Age-related inflammation and insulin resistance: A review of their intricate interdependency[J]. Arch Pharm Res, 2014,37(12):1507-1514.
[8] KUO C C, LIN W T, LIANG C M, et al. Class Ⅰ and Ⅲ phosphatidylinositol 3'-Kinase play distinct roles in TLR signaling pathway[J]. J Immunol, 2006,176(10):5943-5949.
[9] SCHMIDT A M, STERN D M. Hyperinsulinemia and vascular dysfunction: the role of nuclear factor-kappaB, yet again[J]. Circ Res, 2000,87(9):722-724.
[10] MITROULIS I, KAMBAS K, RITIS K. Neutrophils, IL-1beta, and gout: Is there a link[J]. Semin Immunopathol, 2013,35(4):501-512.
[11] AMARAL F A, COSTA V V, TAVARES L D, et al. NLRP3 inflammasome-mediated neutrophil recruitment and hypernociception depend on leukotriene B(4) in a murine model of gout[J]. Arthritis Rheum, 2012,64(2):474-484.
[12] TORRES R, MACDONALD L, CROLL S D, et al. Hyperalgesia, synovitis and multiple biomarkers of inflammation are suppressed by interleukin 1 inhibition in a novel animal model of gouty arthritis[J]. Ann Rheum Dis, 2009,68(10):1602-1608.
[13] CUMPELIK A, ANKLI B, ZECHER D, et al. Neutrophil microvesicles resolve gout by inhibiting C5a-mediated priming of the inflammasome[J]. Ann Rheum Dis, 2016,75(6):1236-1245.
[14] WEICHHART T, COSTANTINO G, POGLITSCH M, et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response[J]. Immunity, 2008,29(4):565-577.
[15] WEICHHART T, HENGSTSCHLAGER M, LINKE M. Regulation of innate immune cell function by mTOR[J]. Nat Rev Immunol, 2015,15(10):599-614.
[16] LEE P S, WILHELMSON A S, HUBNER A P, et al. mTORC1-S6K activation by endotoxin contributes to cytokine up-regulation and early lethality in animals[J]. PLoS One, 2010,5(12): e14399.
[17] PERKINS N D. Integrating cell-signalling pathways with NF-kappa B and IKK function[J]. Nat Rev Mol Cell Bio, 2007,8(1):49-62.
[18] MATSUBARA K, MATSUZAWA Y, JIAO S, et al. Relationship between hypertriglyceridemia and uric acid production in primary gout[J]. Metabolism, 1989,38(7):698-701.
[19] ZHOU Y, FANG L, JIANG L, et al. Uric acid induces renal inflammation via activating tubular NF-kappaB signaling pathway[J]. PLoS One, 2012,7(6): e39738.
[20] MCLAUGHLIN T, LIU L F, LAMENDOLA C, et al. T-cell profile in adipose tissue is associated with insulin resistance and systemic inflammation in humans[J]. Arterioscler Thromb Vasc Biol, 2014,34(12):2637-2643.
猜你喜歡
人人健康(2023年26期)2023-12-07 03:55:46
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
火花(2019年12期)2019-12-26 01:00:28
中國生殖健康(2019年2期)2019-08-23 08:12:10
人大建設(2019年12期)2019-05-21 02:55:32
學苑創造·A版(2015年11期)2016-01-14 09:03:27
中國衛生標準管理(2015年1期)2016-01-14 03:41:27
藥學與臨床研究(2015年4期)2015-06-05 11:35:51
中國醫藥科學(2015年15期)2015-02-27 12:32:27
中國全科醫學(2013年36期)2013-01-25 06:20:58
