PtRu合金催化劑電催化氧化甲醇的作用機理
2018-05-30 09:02:05王媛媛原沁波衛國強段東紅張忠林王恩志郝曉剛劉世斌
太原理工大學學報 2018年3期
關鍵詞:催化劑
王媛媛,原沁波,衛國強,段東紅,張忠林,王恩志,李 瑜,郝曉剛,劉世斌
(1.太原理工大學 化學化工學院,太原 030024;2.清華大學 水沙科學與水利水電工程國家重點實驗室,三江源協同創新研究中心,北京 100084)
直接甲醇燃料電池(DMFC)結構簡單,操作簡單,安全環保,易于運輸,應用前景十分廣闊,已成為21世紀最重要的綠色化學與實現清潔能源的新技術之一[1]。納米金屬Pt是現今DMFC單元催化劑中活性最高、使用最為普遍的材料,但DMFC陰陽兩極的電化學反應屬于多相催化過程,反應中間產物(COads等)易引起催化劑中毒失活[2],以至于納米金屬Pt性能低下和失活成為DMFC研究發展的關鍵核心問題。催化機理的明確與深化理解對催化劑的發展至關重要[3],催化機理的研究可為提高催化劑性能,改進催化劑制備方法提供堅實的理論依據。
PtRu合金是目前穩定性以及電化學活性最好的一種納米合金催化劑[4],但對于其電催化甲醇氧化機理一直存在爭議,阻礙了Pt基納米金屬催化劑的進一步發展。據已有的文獻可知,較多的研究者主張“雙功能機理”[5-7]。這一機理早在1987年由WATANABE et al[8]在研究PtRu納米合金電催化氧化甲醇的過程中提出。機理認為,H2O分子在PtRu納米合金的表面Ru活性位上發生活化,活化生成的OHads可促進PtRu納米合金在低電勢下氧化占據Pt活性位的COads,從而減少COads對Pt活性位的覆蓋率,提高抗COads中毒的性能。電子效應[4,9]是TODA et al[10]在合成并研究PtFe納米合金催化劑性能時提出。研究發現,當Fe原子比達到50%時,其催化活性比納米Pt/C高出25倍,認為活性提高的主要原因是納米金屬表面Pt原子的5d軌道空穴的增加,不是Pt原子間距的改變,由此提出了一個新的理論即“電子效應機理”。之后,LIU et al[11]合成了SiO2負載的PtPd納米合金,在對比XPS測試的納米合金與納米純Pt的Pt4f結合能時發現,合金中Pt4f結合能都向更大值偏移,說明PtPd納米合金中Pt原子與Pd原子間存在強烈的電子交互作用。
核殼結構催化劑中,殼層金屬會與核層金屬之間發生相互作用(如電子效應、協同效應等),從而改變殼層金屬性質,提高催化性能。目前,成功合成的核殼型催化劑有:Fe2O3@Pt,Ag@Pt,Co@Pt,Ni@Pt,Cu@Pt,Ru@Pt[12]等。RAMREZ et al[13]采用量子化學法研究二元核殼型R@Pt(R=Au,Rh,Co,Cu,Ni,Ag等)催化劑模型ORR活性時,通過計算發現,核層金屬可改變殼層Pt原子的d軌道空穴和Pt晶格間距,有效減弱CO在Pt原子表面的吸附,提高氧還原反應轉換效率,說明雙金屬核殼結構催化劑中電子交互作用提高其催化活性。
目前,眾多研究者們或主張雙功能機理,或主張電子效應機理,對Pt基納米合金催化活性提升的認識不夠一致,明顯影響了新型催化劑的研究發展進程。雖然,雙功能機理和電子效應從不同的角度出發,能夠闡明Pt基納米合金的一些實驗現象,各有一定的優勢,但都存在某些實驗現象無法解釋的困境。
本課題組趙碩等[14]成功合成了Ru@Pt/C核殼型催化劑,并且證明其存在電子效應。本文在此基礎上采用不同的制備方法合成直徑相近、Pt/Ru原子比例不同的合金型PtRu/C和核殼型Ru@Pt/C催化劑,通過EDS、XPS等一系列物理技術手段,不同的表征方式說明兩種催化劑中Pt,Ru原子間的電子效應的存在及其強度;通過循環伏安法、交流阻抗法表征催化劑電化學性能,通過對比分析,進一步揭示明確合金型PtRu/C催化劑電催化甲醇氧化的催化作用機理,為DMFC用Pt基納米金屬催化劑的發展提供支持。
1 實驗部分
1.1 材料與試劑
四氯化鉑(PtCl4,99.9%,ALDRICH),三氯化釕(RuCl3,99.9%,ALDRICH),硼氫化鈉(NaBH4,99.99%,ALDRICH),Nafion乳液(質量分數5%,美國杜邦公司);XC-72導電炭黑(美國Carbor公司),其余試劑均為分析純級。
1.2 催化劑的制備
PtRu/C、Pt/C催化劑的制備采用文獻[15]制備方法。先將PtCl4、RuCl3前驅體溶解于四氫呋喃(THF)、超純水(H2O)、乙醇(C2H5OH)三元混合溶液中,負載在XC-72導電碳黑上,再以NaBH4溶液作還原劑充分還原,離心洗滌后,真空烘干,研磨制得催化劑。
Ru@Pt/C催化劑的制備采用KAPLAN et al[16]所發表文獻中制備方法。RuCl3溶解于HCl溶液中,XC-72導電碳黑負載后,調節PH值,分兩次滴加NaBH4還原劑,之后再加入PtCl4的HCl溶液,滴加NaBH4還原劑。最后離心洗滌,真空烘干,研磨制得催化劑。
不同n(Pt)∶n(Ru)比例的樣品可通過調整PtCl4,NaBH4和XC-72導電碳黑等的加入量用上述方法制備,純Pt/C則無需加入RuCl3,依據Pt/Ru原子比,分別將樣品標記為Pt0.33Ru1/C,Pt0.5Ru1/C,Pt1Ru1/C,Pt2Ru1/C,Pt/C;Ru1@Pt0.33/C,Ru1@Pt0.5/C,Ru1@Pt1/C,Ru1@Pt2/C.
1.3 薄膜工作電極的制備
以玻碳電極作為工作電極基底,用超細Al2O3粉末(<2 μm)擦拭玻碳電極表面,之后放在丙酮中超聲洗滌去除表面雜質,放入烘箱空氣氣氛下40 ℃烘干;稱量0.012 g樣品于10 mL燒杯中,加入3 mL超純水、2 mL無水乙醇混合,細胞粉碎機中超聲分散10 min至混合物形成油墨狀,微量移液器量取25 μL懸濁液涂于玻碳電極表面,空氣氣氛下烘箱中50 ℃烘干,再取10 μL Nafion乳液涂催化劑上,紅外燈下80 ℃烘烤成膜,即得到薄膜工作電極。本實驗工作電極催化劑擔載量為0.212 mg/cm2.
1.4 催化劑的物理表征
催化劑晶型采用D/max-2500型X射線衍射儀(XRD,日本Rigaku公司)進行檢測,輻射源為Cu Kα,管電壓40 kV,管電流40 mA,掃描范圍2θ=5°~85°,掃描速度8(°)/min.催化劑的微觀形貌采用JEM-2010型透射電子顯微鏡(TEM,日本電子株氏會社)進行觀察,加速電壓為200 kV.催化劑元素組成采用LEO 438VP型能譜分析儀(EDS,德國 LEO公司)進行測量。納米金屬淺表面層元素組成及電子態采用Thermo Scientific ESCALAB 250Xi-XL型X射線光電子能譜儀(XPS,美國熱電公司)進行分析。
1.5 催化劑的電化學表征
用三電極體系在VMP2型多通道恒電位儀(美國PAR公司)上對催化劑樣品進行電化學性能表征。其中玻碳電極(中國艾達恒晟科技有限公司)為工作電極,Pt片(中國高仕睿聯科技有限公司)為對電極,Hg/Hg2SO4(MSE,中國艾達恒晟科技有限公司)為參比電極;電解液為0.5 mol/L H2SO4+xmol·L-1CH3OH(x=0,0.2,0.5,1.0),實驗均在室溫(25±1 ℃)下進行;循環伏安測試電位區間為-0.74 V~0.70 V,掃速為20 mV/s;交流阻抗測試的測試電位為0.2 V.測試前,電解液中通入高純Ar氣約30 min,除去電解液中溶解的O2,CO2.
2 結果與討論
2.1 催化劑的物理表證結果
PtRu/C和Pt/C催化劑的XRD譜圖見圖1(a).譜圖中A,B,C樣品存在4個較強的衍射峰,衍射角2θ=39.76°,46.24°,67.45°,81.29°分別對應于Pt的(111)、(200)、(220)及(311)晶面,未出現金屬Ru的衍射峰,說明Ru原子可能以合金形式存在,Pt基納米合金呈面心立方晶體結構;而且隨著Pt原子比的減小,衍射峰強度減弱,表明納米金屬粒子的晶化程度有降低的趨勢。在Pt0.5Ru1/C催化劑的譜圖中(D),Pt(200),(220),(322)晶面的衍射峰幾乎消失,Pt(111)晶面衍射峰出現寬化,這可能是因為Pt原子比較小,晶型不夠完整,有非晶態的趨勢;在Pt0.33Ru1/C催化劑的譜圖中(E),僅在2θ=39.76°位置出現了一個寬的禿峰,這可能是由于Pt原子比過小,Pt原子的配位數降低,原子間距增加,形成了類似非晶態的納米粒子,此時Pt0.33Ru1/C的性能不再遵循晶態納米金屬的規律。圖中Pt2Ru1/C(B),Pt1Ru1/C(C),Pt0.5Ru1/C(D)催化劑的Pt(111)晶面衍射峰相對于Pt/C較明顯地向右偏移,這一現象表明,Ru原子進入金屬Pt的晶格,取代了部分Pt原子,形成PtRu合金[17]。

A-Pt/C;B-n(Pt)∶n(Ru)=2∶1;C-n(Pt)∶n(Ru)=1∶1;D-n(Pt)∶n(Ru)=0.5∶1;E-n(Pt)∶n(Ru)=0.33∶1圖1 不同n(Pt)∶n(Ru)比值的PtRu/C和Ru@Pt/C催化劑的XRD譜圖Fig.1 XRD spectra of PtRu/C and Ru@Pt/C catalysts with different n(Pt)∶n(Ru) values
Ru@Pt/C和Pt/C催化劑的XRD譜圖見圖1(b).譜圖中衍射角2θ=39.76°,46.24°處的衍射峰分別對應于Pt(111),(200)晶面,未出現金屬Ru的衍射峰,初步推斷金屬Ru核層被金屬Pt形成的殼層包裹,形成核殼型Ru@Pt/C催化劑。對比圖中Ru1@Pt0.33/C,Ru1@Pt0.5/C,Ru1@Pt1/C,Ru1@Pt2/C和Pt/C的Pt(111)晶面衍射峰發現,隨著Pt原子比的增大,衍射峰強度增強。Ru1@Pt0.33/C(E)催化劑Pt(111)衍射峰強度較低且寬化,其它晶面的衍射峰幾乎觀察不到,可能是因為Pt原子比過小,Pt殼層過薄,呈現為微晶薄膜結構[19];Ru1@Pt0.5/C(D)催化劑的Pt(111)晶面衍射峰有明顯的寬化現象,說明金屬Pt殼層的厚度較薄,但在這樣的厚度下金屬Pt殼層已接近單晶結構。
PtRu/C催化劑和Ru@Pt/C催化劑中Pt(111)晶面衍射峰相對于Pt/C發生偏移,是因為Ru原子的加入對Pt原子產生電子誘導效應,改變了Pt金屬晶體的晶格參數[18]。這種情況下,本研究用PtRu/C催化劑和Ru@Pt/C催化劑中Pt(111)晶面衍射峰相對于Pt/C催化劑Pt(111)晶面衍射峰的偏移值表征Pt基納米金屬存在的電子效應強度。不考慮Pt0.33Ru1/C催化劑,由圖2可見,Pt納米金屬呈晶態結構時,隨Pt原子比增大,兩種催化劑Pt(111)晶面衍射峰偏移值減小,即電子效應強度減小,在各自體系中,Pt1Ru1/C,Ru1@Pt0.5/C催化劑電子效應最強,Ru1@Pt0.5/C強度大于Pt1Ru1/C催化劑。n(Pt)∶n(Ru)=1∶1時,兩種催化劑電子效應接近,n(Pt)∶n(Ru)=2∶1時,Ru@Pt/C催化劑電子效應大于PtRu/C催化劑,這是因為Pt,Ru金屬形成核殼結構,當Pt原子比較大時,Ru金屬核可通過晶格膨脹效應補償,使金屬Pt殼層的晶格參數變化幅度相對較大。當Pt金屬有非晶態趨勢時,Pt原子的配位數有所降低,也不再嚴格遵循晶態結構金屬Pt的規律,所以Pt0.5Ru1/C催化劑的電子效應減弱。
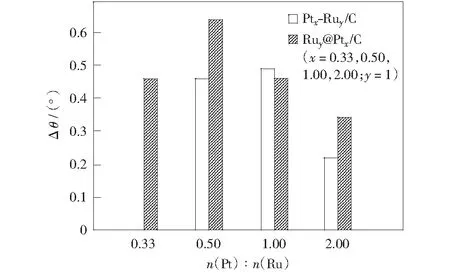
圖2 PtRu/C、Ru@Pt/C催化劑的Pt(111)晶面衍射峰相對于Pt/C偏移值Δθ與n(Pt)∶n(Ru)比值的關系Fig.2 Histogram between Δθ of specific Pt (111) diffraction peak and different n(Pt)∶n(Ru) values of PtRu/C,Ru@Pt/C catalysts
表1和表2分別為制備的PtxRuy/C和Ruy@Ptx/C兩類催化劑的EDS、XPS測試結果。EDS檢測到的Pt/Ru原子比可近似為納米金屬體相的平均原子比,而XPS檢測得到的Pt/Ru原子比值是納米金屬淺表層原子的平均原子比[20]。由表1和表2中EDS數據可看到,EDS實測出的Pt/Ru原子比與加入前驅體鹽Pt/Ru原子比計算值相近,表明在適宜的實驗操作條件下NaBH4能夠將PtCl4和RuCl3完全還原。表1中XPS實測出的Pt/Ru原子比也與加入前驅體鹽Pt/Ru原子比計算值近似相同,這說明本研究制備的合金型PtRu/C催化劑確實為合金結構。表2中XPS測出的Pt/Ru原子比遠大于同一樣品EDS測出的Pt/Ru原子比,說明本研究制備的核殼型Ru@Pt/C催化劑納米金屬表面擁有更高比例的Pt元素,結合本研究制備方法的特點,進一步證明納米金屬Ru核被納米金屬Pt層包裹,Ru@Pt/C催化劑呈核殼型納米結構[21]。
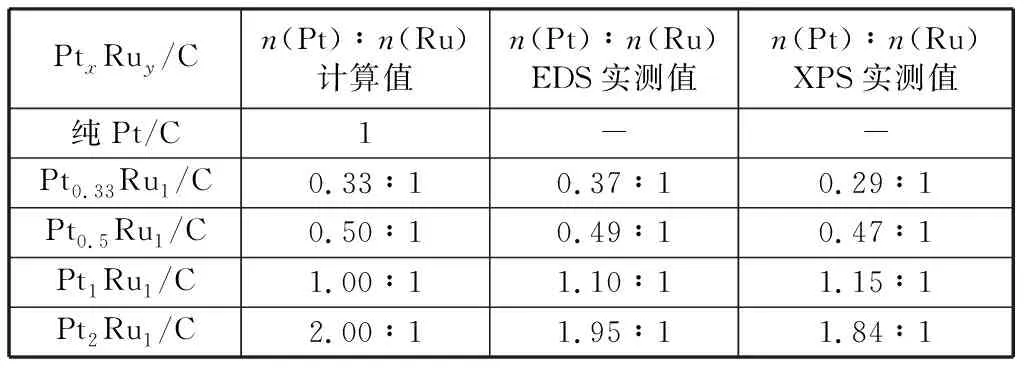
表1 不同Pt/Ru原子比PtxRuy/C催化劑n(Pt)∶n(Ru)計算值及EDS,XPS實測值Table 1 Comparisons between stoichiometric value of n(Pt)∶n(Ru) and EDS,XPS measured value of n(Pt)∶n(Ru) in different PtxRuy/C catalysts

表2 不同Pt/Ru原子比Ruy@Ptx/C催化劑n(Pt)∶n(Ru)計算值及EDS,XPS實測值Table 2 Comparisons between stoichiometric value of n(Pt)∶n(Ru) and EDS,XPS measured value of n(Pt)∶n(Ru) in different Ruy@Ptx/C catalysts
圖3展示了不同n(Pt)∶n(Ru)比值PtRu/C和Ru@Pt/C催化劑中Pt4f軌道的結合能。圖中,兩種催化劑中的Pt4f結合能相對于納米純Pt的Pt4f結合能均發生正移,此現象說明合金和核殼結構中Pt原子的電子向Ru原子未填滿的d軌道轉移[11]443,Pt的5d軌道空穴增加,內層軌道電子的結合能增大。通常,這種Pt原子和Ru原子的電子交互作用,被稱為“電子效應”。
圖4為兩種催化劑的Pt4f7/2結合能相對于Pt/C催化劑Pt4f7/2結合能的偏移值與Pt/Ru原子比的關系。可看出當Pt呈晶態結構時,兩種催化劑隨著Pt原子比的增大Pt4f7/2結合能偏移值減小,即電子效應強度減弱, Pt1Ru1/C和Ru1@Pt0.5/C催化劑在各自體系中電子效應最強,且Ru1@Pt1,2/C催化劑分別與 Pt1Ru1,2/C催化劑的電子效應強度相近。當納米Pt呈非晶態結構或有非晶態趨勢時,Pt0.5Ru1/C,Pt0.33Ru1/C和Ru1@Pt0.33/C催化劑的Pt4f7/2結合能偏移值減小,不具備晶態結構Pt金屬的性質。

A-n(Pt)∶n(Ru)=1∶1;B-n(Pt)∶n(Ru)=2∶1;C-Pt/C;D-n(Pt)∶n(Ru)=0.5∶1;E-n(Pt)∶n(Ru)=0.33∶1圖3 不同n(Pt)∶n(Ru)比值PtRu/C催化劑和Ru@Pt/C催化劑Pt4f軌道的精細譜圖Fig.3 Spectrogram of Pt4f orbits of different PtRu/C and Ru@Pt/C catalysts
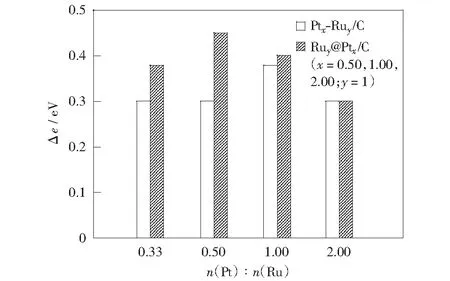
圖4 不同催化劑的Pt4f7/2結合能偏移值Δe與n(Pt)∶n(Ru)比值的關系Fig.4 Diagram between Δe of Pt4f7/2 binding energy peaks and different n(Pt)∶n(Ru) values of different catalysts
Pt1Ru1/C催化劑的HRTEM見于圖5.不同Pt/Ru比例的PtRu/C催化劑制備方法相同,所以總體形貌和結構類似,在此選取Pt1Ru1/C催化劑電鏡圖進行分析。從圖5(a)可知,制備出的催化劑呈球形或類球形,分散性較好。由圖5(b)可看出金屬粒子的晶體結構,平均粒徑約為9~11 nm,局部放大圖中不同金屬粒子各自晶格的規則明顯、走向不同,表明Pt原子與Ru原子形成了合金型催化劑。
核殼型Ru1@Pt0.5/C催化劑的HRTEM見于圖6.選取Ru1@Pt0.5/C催化劑電鏡圖進行分析。從圖6(a)可知,催化劑呈球形或類球形,分散性較好。由圖6(b)可看出金屬粒子的晶體結構,中間顏色較深的為Ru核層金屬,外面顏色較淺的殼層為Pt金屬,厚度約為1~2 nm,平均粒徑約10 nm,與合金型PtRu/C催化劑粒徑相近。

圖5 Pt1Ru1/C催化劑的HRTEM圖Fig.5 HRTEM images of Pt1Ru1/C alloy catalysts

圖6 Ru1@Pt0.5/C催化劑的HRTEM圖Fig.6 HRTEM images of Ru1@Pt0.5/C core-shell catalysts
2.2 催化劑的電化學表證結果
圖7展示了3種催化劑在0.5 mol/L H2SO4溶液中的循環伏安曲線,金屬Pt的吸脫附CV曲線的變化可分為4個電位區間,即-0.75~-0.4 V氫的吸脫附區,-0.4~-0.2 V的雙電層區和-0.2~0.6 V的氧化區和高于0.6 V的析氧區。圖中可看出,Ru@Pt/C展示與Pt/C催化劑非常相似的氫吸脫附和雙電層的CV特征,并未出現金屬Ru的溶解氧化峰,在-0.2~0.6 V氧化區和析氧區顯示出較高的氧化電流,在回掃過程中,氧還原電流明顯較高,表明本研究制備的Ru@Pt/C催化劑易于被氧化,對析氧反應、氧還原反應的活性較高,符合核殼結構特征[22]。PtRu/C催化劑的氧化電流峰較其他兩種催化劑有一定提前,各區的反應電流相應較大,具有PtRu/C催化劑的特征[23]。
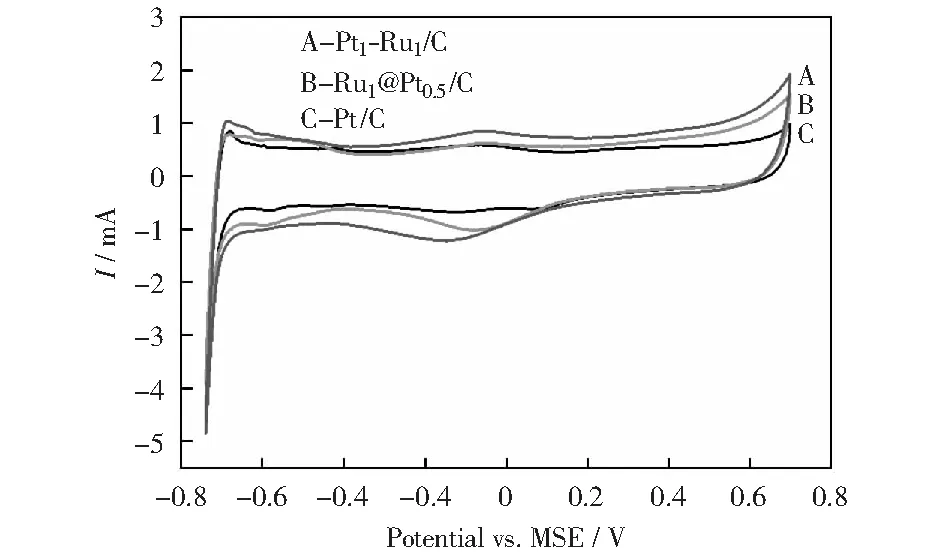
圖7 不同催化劑在0.5 mol/L H2SO4溶液中的循環伏安曲線Fig.7 CV curves of different catalysts in 0.5 mol/L H2SO4

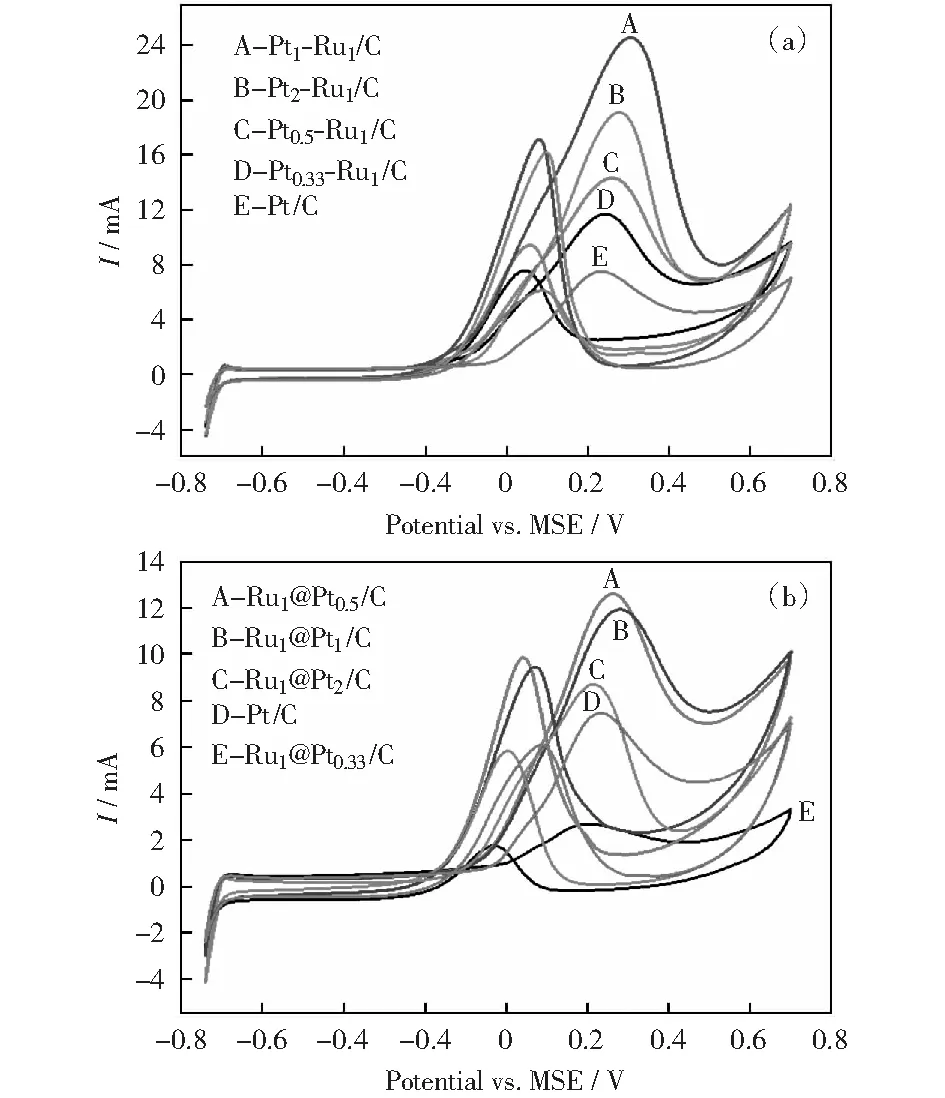
圖8 不同n(Pt)∶n(Ru)比值PtRu/C和Ru@Pt/C催化劑在0.5 mol/L H2SO4+1.0 mol/L CH3OH溶液中的循環伏安曲線Fig.8 CV curves of different PtRu/C and Ru@Pt/C catalysts in 0.5 mol/L H2SO4+1.0 mol/L CH3OH
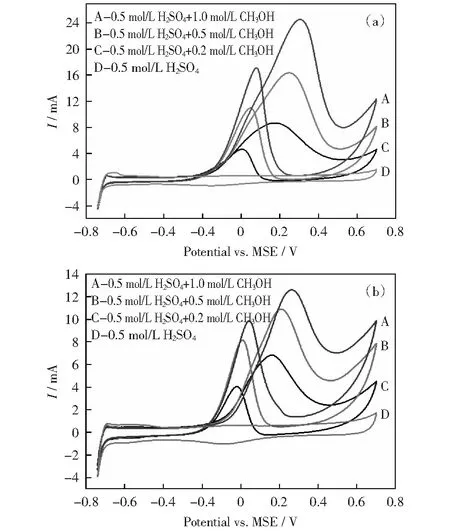
圖9 Pb1Ru1/C和Ru1@Pt0.5/C催化劑在不同甲醇濃度電解液中的循環伏安曲線Fig.9 CV curves of Pb1Ru/C and Ru1@Pt0.5/C catalysts in electrolytes with different methanol concentration
圖10比較了不同催化劑在0.5 mol/L H2SO4+1.0 mol/L CH3OH電解液中甲醇氧化峰電流、交流阻抗值與Pt/Ru原子比的關系。從圖中可看出,隨Pt/Ru原子比的增大,Ru@Pt/C催化劑催化活性先增大后減小,Ru1@Pt0.5/C的活性最高,此規律與其電子效應對Pt/Ru原子比關系的規律相一致。Ru1@Pt0.33/C盡管也存在類似于其它核殼型納米金屬的電子效應,但催化活性低于Pt/C,說明該催化劑可能存在其它的催化效應,其關鍵在于Pt原子比較小,Pt殼層呈多微晶結構,晶界原子的比例較高,相應的催化機理有待進一步探討。
相對于Ru@Pt/C催化劑,同一Pt/Ru比例下,PtRu/C催化劑電催化甲醇的峰電流均較高,比較兩種類型催化活性最高的催化劑,Pt1Ru1/C的峰電流約為Ru1@Pt0.5/C的2倍。圖10(b)中兩種催化劑的交流阻抗特性均呈“倒火山型”,所反映出的催化活性的規律與循環伏安法的規律相一致。兩種電化學方法的測試結果說明,合金型PtRu/C催化劑電催化活性明顯優于核殼型Ru@Pt/C催化劑。
圖11(a)是在0.5 mol/L H2SO4+1.0 mol/L
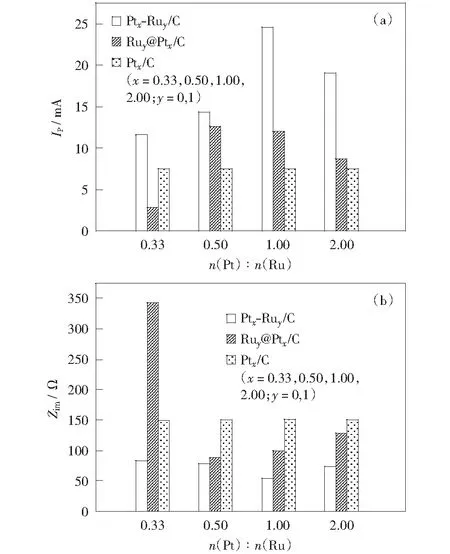
圖10 不同催化劑在0.5 mol/L H2SO4+1.0 mol/L CH3OH電解液中的峰電流與n(Pt)∶n(Ru)比值的關系圖(a)和電荷轉移電阻與n(Pt)∶n(Ru)比值的關系圖(b)Fig.10 Diagram of the peak current vs. n(Pt)∶n(Ru) (a) and the impedance spectra vs. n(Pt)∶n(Ru) (b) of different catalysts in 0.5 mol/L H2SO4+1.0 mol/L CH3OH
CH3OH中,Ru@Pt/C催化劑催化甲醇氧化峰電流相對于Pt/C的增加值與Pt4f軌道電子結合能偏移值(作為電子效應的表征值)的關系曲線。圖中峰電流增加值即為Ru@Pt/C在表面Pt原子電子效應作用下相對于Pt/C所增加的催化活性,由圖中的關系曲線可見,Ru@Pt/C的催化活性增加幅度與電子效應強度基本呈線性關系。這里Ru1@Pt0.33/C中Pt呈微晶態薄膜,不遵循晶態金屬Pt規律,不參與線性擬合。由XPS測試結果可知,Pt1Ru1/C,Pt2Ru1/C中Ru的3p1軌道結合能分別相對純金屬Ru原子下降3.7,3.9 eV,說明PtRu納米合金中Ru原子接收到電子,此效應與合金中Pt原子移去電子相印證。在PtRu納米合金的晶格完整情況下,其表面Pt,Ru原子所占面積比與其原子比相近,如Pt1Ru1表面Pt,Ru原子所占面積各為1/2.假設表面Ru原子所具催化活性與Pt原子相同,并且假設因電子效應導致的峰電流增加值符合圖11(a)中曲線關系,則Pt1Ru1/C表面Pt,Ru原子因電子效應導致的活性提升幅度(以峰電流提升值表征)約為3.53 mA,圖11(b)中分別列出了Pt/C催化甲醇氧化峰電流值、電子效應促進作用下PtRu/C的峰電流計算值、以及PtRu/C催化甲醇氧化峰電流實測值。可看出,即使假設Ru原子所具活性與Pt原子相同,PtRu/C催化甲醇氧化峰電流實測值依然遠大于有電子效應促進作用的計算值。眾所周知,金屬Ru對甲醇電氧化的活性遠遠低于金屬Pt,PtRu/C納米合金催化劑的活性相對于Pt/C大幅提高的原因,不僅僅限于納米合金中的電子效應,還應該與Ru原子活性位的特殊性能有關。

圖11 催化劑在0.5 mol/L H2SO4+1.0 mol/L CH3OH電解液中峰電流與電子效應的關系圖(a)以及峰電流計算值、實測值與n(Pt)∶n(Ru)比值的關系圖(b)Fig.11 Relation curves of the peak current vs. electronic effect (a) and relation curves of peak current vs. n(Pt)∶n(Ru) (b) of PtRu/C catalyst in 0.5 mol/L H2SO4+1.0 mol/L CH3OH
根據WILLIAM et al[26]的研究可以知道,甲醇在金屬Pt電極表面的電氧化有以下主要步驟:

由公式可知,甲醇通過一系列脫氫和吸附解離步驟實現氧化,其中中間產物COads占據Pt表面活性位,易引起催化劑中毒。雙功能機理認為:Ru原子的特性在于,它不吸附甲醇[26],只吸附H2O分子,可快速在Ru原子表面形成OHads,Ru-OHads促進CO在低電勢下氧化,因此大幅度提升了催化劑的抗CO中毒能力和催化活性,如下公式所示:

至此可認為峰電流實測值高出計算值的部分是雙功能機理作用下的催化活性提高。綜合以上不同方面的信息可以確信,合金型PtRu/C催化劑電催化甲醇氧化是電子效應和雙功能機理共同作用。
3 結論
1) 制備的3種納米金屬粒子為球形,平均粒徑較為接近,約10 nm,物理表征和電化學證實,Ru@Pt納米金屬為核殼型結構,PtRu納米金屬為合金型結構。
2) 兩種雙金屬納米粒子的Pt4f結合能都有增大趨勢,表明Pt原子的電子向Ru原子的d軌道偏移,增加了Pt5d軌道空穴。這種不同元素原子間的電子偏移,即所謂“電子效應”,促進了Ru@Pt/C對甲醇電氧化的催化活性,相較Pt/C有明顯提高,而且電子效應越強,納米金屬Pt的活性越高。
3) 不同電化學方法表征結果表明,合金型PtRu/C催化劑電催化甲醇氧化的活性明顯高于Ru@Pt和Pt/C,其催化作用機制不僅存在Pt活性位的“電子效應”促進作用,而且還存在Pt,Ru活性位的“雙功能活性中心”促進作用。有關PtRu納米合金催化機制的認識,將對Pt基納米金屬新型催化劑的研發提供新的基礎。
:
[1] 李金峰,宋煥巧,邱新平.直接甲醇燃料電池陽極催化劑的研究進展[J].電源技術,2007,131(2):167-170.
LI J F,SONG H Q,QIU X P.Research progress of anode catalysts for direct methanol fuel cell[J].Chinese Journal of Power Sources,2007,131(2):167-170.
[2] SONG S,ZHOU W,LIANG Z,et al.The effect of methanol and ethanol cross-over on the performance of PtRu/C-based anode DAFCs[J].Applied Catalysis B Environmental,2005,55(1):65-72.
[3] MUNK J,CHRISTENSEN P A,HAMNETT A,et al.The electrochemical oxidation of methanol on platinum and platinum+ruthenium particulate electrodes studied by in-situ FTIR spectroscopy and electrochemical mass spectrometry[J].Journal of Electroanalytical Chemistry,1996,401(1/2):215-222.
[4] 李偉,黃青丹,黃紅良,等.DMFC陽極PtRu催化劑的研究進展[J].電池工業,2007,12(3):200-204.
LI W,HUANG Q D,HUANG H L,et al.Research progress of PtRu catalysts for direct methanol fuel cells[J].Chinese Battery Industry,2007,12(3):200-204.
[5] IWASITA T,HOSTER H,JOHN-ANACKER A,et al.Methanol oxidation on PtRu electrodes.Influence of surface structure and Pt-Ru atom distribution[J].Langmuir,2000,16(2):522-529.
[6] LEE C H,LEE C W,KIM D I,et al.Characteristics of methanol oxidation on Pt-Ru catalysts supported by HOPG in sulfuric acid[J].International journal of hydrogen energy,2002,27(4):445-450.
[7] TING C C,LIU C H,TAI C Y,et al.The size effect of titania-supported Pt catalysts on the electrocatalytic activity towards methanol oxidation reaction primarily via the bifunctional mechanism[J].Journal of Power Sources,2015,280:166-172.
[8] WANTANABE M,UCHIDA M,MOTOO S.Preparation of highly dispersed Pt+Ru alloy clusters and the activity for the electro-oxidation of methanol[J].Journal of Electro-analytical Chemistry and Interfacial Electrochemistry,1987,229(1/2):395-406.
[9] PEREIRA L G S,PAGANIN V A,TICIANELLI E A.Investigation of the CO tolerance mechanism at several Pt-based bimetallic anode electrocatalysts in a PEM fuel cell[J].Electrochimica Acta,2009,54(7):1992-1998.
[10] TODA T,IGARASHI H,WATANABE M.Enhancement of the electrocatalytic O2reduction on Pt-Fe alloys[J].Journal of Electroanalytical Chemistry,1999,460(1):258-262.
[11] LIU J,ZOU S,XIAO L,et al.Well-dispersed bimetallic catalysts confined in mesoporous metal oxides and their optimized catalytic activity for nitrobenzene hydrogenation[J].Catalysis Science & Technology,2014,4(2):441-446.
[12] YANG J,LEE J Y,CHEN L X,et al.A phase-transfer identification of core-shell structures in Ag-Pt catalysts[J].The Journal of Physical Chemistry B,2005,109(12):5468-5472.
[14] 趙碩,段東紅,衛國強,等.核殼型Ru@Pt在甲醇電催化氧化反應中的電子效應研究[J].太原理工大學學報,2016,47(4):471-477.
ZHAO S,DUAN D H,WEI G Q,et al.Electronic effect of core-shell Ru@Pt nano-particle catalysts in methanol electro-oxidation[J].Journal of Taiyuan University of Technology,2016,47(4):471-477.
[15] 陳煜,唐亞文,李鋼,等.一種制備碳載高合金化Pt-Ru催化劑的方法 [J].無機化學學報,2006,22(1):59-64.
CHEN Y,TANG Y W,LI G,et al.Preparation of carbon supported high alloying Pt-Ru catalyst[J].Chinese Journal of Inorganic Chemistry,2006,22(1):59-64.
[16] KAPLAN D,BURSTEIN L,ROSENBERG Y,et al.Comparison of methanol and ethylene glycol oxidation by alloy and core-shell platinum based catalysts[J].Journal of Power Sources,2011,196(20):8286-8292.
[17] ZHANG J M,ZHU F F,ZHANG K H,et al.Pt-Ru catalysts prepared by a modified polyol process for direct methanol fuel cells[J].Precious Metals,2012(s1):222-226.
[18] CAMARA G A,GIZ M J,PAGANIN V A,et al.Correlation of electrochemical and physical properties of PtRu alloy electrocatalysts for PEM fuel cells[J].Journal of Electroanalytical Chemistry,2002,537(1):21-29.
[19] LIU S B,MA Y H,WEI G Q,et al.Methanol electro-oxidation kinetics on Ni@Pt nanoparticles coated by amorphous metal(In Chinese)[J].Journal of Taiyuan University of Technology,2012,43(3):300-304.
[20] BECKER E D,FARRAR T C.Fourier transform spectroscopy[J].Science,1972,178(4059):361-368.
[21] MUTHUSWAMY N,DE LA Fuente J L G,TRAN D T,et al.Ru@Pt core-shell catalysts for methanol fuel cell catalyst:Control and effects of shell composition[J].International Journal of Hydrogen Energy,2013,38(36):16631-16641.
[22] WANG J J,LIU Y T,CHEN I L,et al.Near-monolayer platinum shell on core-shell nanocatalysts for high-performance direct methanol fuel cell[J].The Journal of Physical Chemistry C,2014,118(5):2253-2262.
[23] KRAUSA M,VIELSTICH W.Study of the electrocatalytic influence of Pt/Ru and Ru on the oxidation of residues of small organic molecules[J].Journal of Electroanalytical Chemistry,1994,379(1/2):307-314.
[24] LIN S D,HSIAO T C,CHANG J R,et al.Morphology of carbon supported Pt-Ru electrocatalyst and the CO tolerance of anodes for PEM fuel cells[J].The Journal of Physical Chemistry B,1999,103(1):97-103.
[25] WATANABE M,MOTOO S.Electrocatalysis by ad-atoms:Part III.Enhancement of the oxidation of carbon monoxide on platinum by ruthenium ad-atoms[J].Journal of Electroanalytical Chemistry and Interfacial Electrochemistry,1975,60(3):275-283.
[26] HOLSTEIN W L,ROSENFELD H D.In-situ X-ray absorption spectroscopy study of Pt and Ru chemistry during methanol electrooxidation[J].The Journal of Physical Chemistry B,2005,109(6):2176-2186.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
