以腦干癥狀起病的視神經脊髓炎譜系疾病32例臨床特征及預后分析
2018-05-09 03:16:35趙致慷黃曉曦劉洪波
中風與神經疾病雜志 2018年4期
關鍵詞:癥狀
趙致慷, 彭 靜, 王 芮, 黃曉曦, 劉洪波
視神經脊髓炎(neuromyelitis optica,NMO)是一種嚴重的主要累及視神經和脊髓的自身免疫性中樞神經系統炎性脫髓鞘疾病[1]。自抗水通道蛋白4(aquaporin-4,AQP4)抗體被發現以來,證明其是一種不同于多發性硬化(multiple sclerosis,MS)的獨立疾病實體[2]。2015年,國際NMO診斷小組(the international panel for NMO diagnosis IPND)對2006年Wingerchuk診斷標準進行修訂,提出了最新的診斷標準,將NMO整合入更廣義的NMOSD疾病范疇。 鑒于AQP4的高度特異性及敏感性,IPND進一步對NMOSD進行診斷分層,將其分為AQP4-IgG陽性組和AQP4-IgG陰性組,并制定了相應診斷細節[3]。 同時,NMO的臨床表現形式也由最初的視神經炎、急性脊髓炎,擴展為包括最后區綜合征、其他急性腦干、間腦、大腦綜合征在內的多種表現形式[4~6]。
目前,臨床上大多數患者仍以視神經炎及急性脊髓炎為主要臨床表現,其他核心癥狀較為少見。少數NMOSD患者可以以腦干癥狀起病[5,7],由于早期缺乏視神經炎或脊髓炎的表現,在臨床實踐中很容易誤診漏診,且這些患者的預后及疾病復發情況似乎欠佳。因此,本文對345例已確診NMOSD患者中以腦干癥狀起病的32例進行回顧,總結分析其臨床特征、影像學表現及預后復發情況,以期為臨床診療提供參考。
1 對象和方法
1.1 研究對象 從2010年1月1日-2017年6月1日在鄭州大學第一附屬醫院確診為NNOSD的345例患者中選取以腦干癥狀起病的32例。此32例患者均符合2015年IPND關于NMOSD的最新診斷標準。
1.2 研究方法
1.2.1 資料收集 收集如下臨床資料:性別、發病時年齡、首診科室、臨床表現、臨床復發、免疫治療、血及腦脊液相關實驗室指標、已行AQP4抗體檢測情況、MOG抗體檢測情況、其他自身免疫性抗體檢測情況、脊髓及頭部MRI及擴展殘疾狀態量表(EDSS)評分情況。
1.2.2 資料分析 回顧性總結分析患者的臨床特征及預后、復發情況,需要統計分析的資料,采用SPSS 17.0 軟件進行分析,計數資料比較采用Pearson卡方及校正卡方。P<0.05表示差異有統計學意義。
2 結 果
2.1 臨床特征 32例患者中女性30例,男性2例。發病年齡15~65歲,平均發病年齡(36.50±8.32)歲。32例患者均以腦干癥狀起病,其中22例主要表現為頑固性惡心、嘔吐及呃逆,10例主要表現為頭暈、復視及共濟失調。此外,尚有2例出現飲水嗆咳、吞咽困難癥狀,2例伴有頭痛,1例伴有四肢無力癥狀。32例患者中,首診科室為神經內科17例,消化內科10例,急診科2例,眼科2例,風濕免疫科1例。15例患者早期存在誤診或延誤診療的情況,占46.88%。所有患者明確診斷后,均接受大劑量甲潑尼龍沖擊治療,并在后續治療中服用激素或免疫抑制劑預防復發。
2.2 實驗室檢查
2.2.1 血清學檢查 32例患者血常規、肝腎功、電解質等指標大致正常。4例抗甲狀腺過氧化物酶抗體陽性,2例抗核抗體及抗SSA抗體陽性,1例抗RO50抗體、抗Sm抗體陽性。28例行AQP4抗體檢查的患者中,陽性19例,占67.86%。11例行過MOG抗體檢查的患者中,2例陽性,此2例患者均為AQP4抗體陰性。
2.2.2 腦脊液檢查 21例患者曾于院內行腰椎穿刺術取腦脊液檢查:其中CSF細胞學檢查發現白細胞計數增高13例,占61.90%。CSF電泳檢查發現腦脊液免疫球蛋白IgG升高12例,占57.14%;IgG生成指數升高14例,均值0.994,占66.67%。24 h CSFIgG合成率升高12例 均值18.25,占57.14%。CSF寡克隆電泳陰性20例,陽性1例,陰性占95.24%。
2.3 影像學特征 頭部及脊髓MRI示:病變多位于延髓背側、腦干背側、四腦室及中腦導水管周圍。呈長T1、長T2信號,FLAIR及DWI上呈稍高信號,部分病灶可見斑片狀強化。病灶部位與腦干癥狀相對應,32例中12例病變下延至頸髓,呈長節段線樣損害。其典型影像學表現(見圖1A~D)。
2.4 預后及復發情況 根據患者入院后的EDSS評分將其分為高分組(5.0)和低分組(4.5),分別表示較高和較低的殘疾情況。32例以腦干起病的NMOSD患者中高分組有24例,低分組8例;在剩余的313例患者中,高分組有168例,低分組有145例。以腦干癥狀起病的NMOSD患者的EDSS評分水平明顯高于其他的非腦干癥狀起病的患者,差異有統計學意義(P=0.021<0.05)。
NMOSD的復發誘因有多種,32例患者中自行停用激素及免疫抑制劑復發的有7例,復發前有上呼吸道感染史的5例,情緒波動的3例,過度勞累的3例,此外尚有16例患者無明顯誘因。
在345例已明確診斷NMOSD患者中,以已知總病程中復發次數3次及復發次數兩次分組,32例以腦干癥狀起病的NMOSD患者中復發次數3次的有15例;剩余患者中有83例復發次數3次,經卡方檢驗,差異有統計學意義(P=0.030<0.05)。若以已知總病程中復發次數5次分組,32例患者中有5例,占15.63%;剩余患者中有11例,占3.51%。經校正卡方檢驗,差異明顯,有統計學意義(P=0.008<0.01)。故可認為32例以腦干癥狀起病的NMOSD患者的預后情況較差且更易復發。
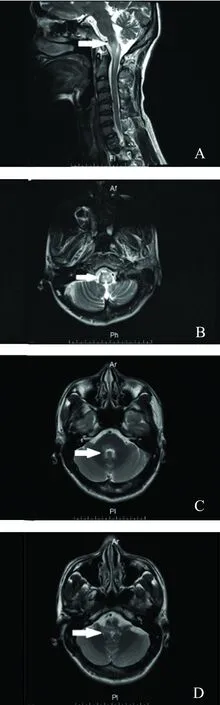
圖1(A~D) 延髓、腦橋及四腦室和中腦導水管周圍長T2信號病灶
3 討 論
NMOSD臨床上多以嚴重的視神經炎和長節段橫貫性脊髓炎為特征表現,常于青壯年起病,女性居多,在亞洲人群中占有較高比例,且為高復發、高致殘性疾病[8,9]。對NMOSD的早期診斷、早期治療、避免誤診、控制復發等都具有重要的臨床意義。
近年來,出現了一些與NMOSD相關的除視神經炎和脊髓炎外的其他臨床表現的相關報道[5~7]。這些臨床綜合征分別累及了除視神經和脊髓外的其他中樞神經區域,包括延髓最后區、腦干及四腦室周邊區、下丘腦區、大腦半球白質及胼胝體區。我們的研究發現,在345例確診NMOSD的患者中有32例以腦干癥狀起病后經影像學證實腦干部位受累,約占總人數的9.3%。另有若干例累及下丘腦區及大腦白質區的特殊病例,因例數較少,未加入分析對比。
NMOSD可以合并腦干癥狀,并且其中的一些患者可以以腦干癥狀起病。Kim W等的一項回顧性研究證實了83例NMOSD患者有7例以腦干癥狀首發;Wang KC等報道在49例NMO患者中,18例存在腦干損害,其中15例以腦干癥狀首發;Kremer L等的一項納入258例患者的多中心研究顯示,有腦干癥狀及腦干損傷的患者在NMO患者中占有相當比例,且在非高加索人中高于高加索人[5,10,11]。
腦干是連接大腦、小腦、脊髓的中間樞紐,是中樞神經系統的核心功能區。多種神經反射中樞及生命活動中樞集中于此,上、下行神經傳導束及神經核團密集,解剖結構復雜[12]。因此,腦干損傷往往病情較重,變化較快,是臨床工作中的難點。
NMOSD腦干病灶通常位于延髓背側、最后區和中腦導水管周圍。頭部及脊髓的MRI檢查通常可發現相應部位存在異常信號。最后區位于第四腦室底部,其內毛細血管缺乏致密的內皮連接,導致血液循環中的自身抗體如AQP4易從此侵入中樞系統。從而引起相應部位腦干損傷,出現惡心、嘔吐、頑固性呃逆等臨床癥狀[13,14]。中腦導水管周圍區及四腦室底,有多處神經核團聚集,包括動眼神經核、滑車神經核、三叉神經核、展神經核、疑核、孤束核等,且小腦上、下腳也與此區毗鄰。此區受累,臨床上可出現頭暈、復視、共濟失調、飲水嗆咳、吞咽困難、頭面痛等多種臨床表現[11,12]。以腦干癥狀起病的NMOSD臨床表現不典型,早期易誤診漏診,造成治療延誤。本研究的32例患者,分別以惡心嘔吐、頑固性呃逆、頭暈、復視、共濟失調等臨床癥狀起病,近半數患者首診于非神經科室,存在誤診及延誤診療的情況。臨床上,對此類以腦干癥狀起病的患者應予以高度重視,各相關科室也應加強相互間的交流、協作,以期對疾病做到早期診斷、規范治療。關于以腦干癥狀起病NMOSD的預后及復發情況,本研究通過EDSS評分及疾病復發次數的比較,證實以腦干癥狀起病的NMOSD患者的預后情況較差且在臨床上更易復發。這可能與腦干部位特殊復雜的解剖結構及功能有一定相關性。
實驗室檢查方面,本研究的患者中多數AQP4抗體陽性,且部分患者合并有相關的自身免疫性抗體,如抗甲狀腺過氧化物酶抗體、抗核抗體、抗SSA抗體等,這與國內外的一些相關報道一致[8,15,16]。在AQP4抗體陰性的患者中,存在2例MOG抗體陽性,提示該抗體可能參與NMOSD的致病,并為AQP4抗體陰性NMOSD的診斷提供一定參考[17,18]。腦脊液檢查發現,多數患者白細胞有升高,免疫球蛋白及IgG生成指數也有明顯升高,大多數病例CSF寡克隆電泳陰性。腦脊液檢查對NMOSD的診斷及鑒別診斷有一定程度的提示意義。
綜上所述,本研究發現,以腦干癥狀起病的NMOSD臨床常以惡心嘔吐、頑固性呃逆、頭暈、復視、共濟失調等非NMO典型癥狀為首發表現,早期易誤診漏診,造成診療延誤。通過比較相關數據,發現此類患者的預后情況也相對較差,且在以后的病程中,復發次數也更頻繁。因此,臨床對此類患者應予以高度重視,盡可能做到早期診斷、規范治療。在其病情緩解后,仍應采取各種手段做好疾病復發的預防,如堅持使用激素及免疫抑制劑,做好患者的病情教育及隨訪,以期降低此類患者的復發率和致殘率。
[參考文獻]
[1]Jarius S,Wildemann B.The history of neuromyelitis optica[J].J Neuroinflam-mation,2013,10(8):1742-2094.
[2]Lennon VA,Wingerchuk DM,Kryzer TJ,et al.A serum autoantibody marker of neuro- myelitis optica:distinction from multiple sclerosis[J].Lancet,2004,364 (9451):2106-2112.
[3]Wingerchuk DM,Banwell B,Bennett JL,et al.International consensus diagnostic criteria for neuromyelitis optica spectrum disorders[J].Neurology,2015,85(2):177-189.
[4]Nagaishi A,Takagi M,Umemura A,et al.Clinical features of neuromyelitis optica in a large Japanese cohort:comparison between phenotypes[J].J Neurol Neurosurg Psychiatry,2011,82(12):1360-1364.
[5]Kim W,Kim SH,Lee SH,et al.Brain abnormalities as an initial manifestation of neuromyelitis optica spectrum disorder[J].Mult Scler,2011,17(9):1107- 1112.
[6]Kim JE,Kim SM,Ahn SW,et al.Brain abnormalities in neuromyelitis optica[J].J Neurol Sci,2011,302(1~2):43-48.
[7]Apiwattanakul M,Popescu BF,Matiello M,et al.Intractable vomiting as the initial presentation of neuromyelitis optica[J].Ann Neurol,2010,68(5):757-761.
[8]Pereira WL,Reiche EM,Kallaur AP,et al.Epidemiological,clinical,and immunological characteristics of neuromyelitis optica:A review[J].Journal of Neurological Sciences,2015,355(1~2):7-17.
[9]Ochi H,Fujihara K.Demyelinating diseases in Asia[J].Curr Opin Neurol,2016,29(3):222-228.
[10]Wang KC,Lee CL,Chen SY,et al.Prominent brainstem symptoms/signs in patients with neuromyelitis optica in a Taiwanese population[J].J Clin Neurosci,2011,18(9):1197-1200.
[11]Kremer L,Mealy M,Jacob A,et al.Brainstem manifestations in neuromyelitis optica:a multicenter study of 258 patients[J].Mult Scler,2014,20(7):843-847.
[12]Hurley RA,Flashman LA,Chow TW,et al.The brainstem:anatomy,assessment,and clinical syndromes[J].J Neuropsychiatry Clin Neurosci,2010,22(1):1-7.
[13]Takahashi T,Miyazawa I,Misu T,et al.Intractable hiccup and nausea in neuromyelitis optica with anti-aquaporin-4 antibody: a herald of acute exacerbations[J].J Neurol Neurosurg Psychiatry,2008,79(9):1075-1078.
[14]Price CJ,Hoyda TD,Ferguson AV,et al.The area postrema: a brain monitor and integrator of systemic autonomic state [J].Neuroscientist,2008,14(2):182-194.
[15]Park JH,Hwang J,Min JH,et al.Presence of anti-Ro/SSA antibody may be associated with anti-aquaporin-4 antibody positivity in neuromyelitis optica spectrum disorder[J].J Neurol Sci,2015,348(1~2):132-135.
[16]Wang X,Yi H,Liu J,et al.Anti-thyroid antibodies and thyroid function in neuromyelitis optica spectrum disorders[J].J Neurol Sci,2016,366:3-7.
[17]Hoftberger R,Sepulveda M,Armangue T,et al.Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease[J].Mult Scler,2015,21(7):866-874.
[18]Uzawa A,Mori M.Seronegative neuromyelitis optica spectrum disorder patients diagnosed using new diagnostic criteria[J].Mult Scler,2016,22(10):1371-1375.
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中老年保健(2021年12期)2021-08-24 03:30:44
今日農業(2020年17期)2020-10-27 03:10:52
今日農業(2020年16期)2020-09-25 03:05:08
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
吉林蔬菜(2017年10期)2017-11-01 07:47:04
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國醫學影像學雜志(2015年9期)2015-12-15 11:03:26
