朗格漢斯細胞組織細胞增生癥2例分析
2018-04-24 03:29:25羅專波黃小萍徐寧李晨蔚樸正華
浙江醫學 2018年6期
羅專波 黃小萍 徐寧 李晨蔚 樸正華
朗格漢斯細胞組織細胞增生癥(Langerhans cell histiocytosis,LCH)是一類罕見的疾病,以組織單克隆朗格漢斯細胞增生、浸潤、肉芽腫形成,導致器官功能障礙為主要臨床特征。根據器官受累的范圍,LCH分為單個器官受累、多器官受累和多系統受累。LCH的發病機制、病理生理過程、治療和預后仍有待于進一步闡明。現將2016年3月至2017年3月寧波市第一醫院呼吸內科收治的2例LCH患者的臨床資料進行報道,并復習有關LCH的研究進展,以提高對本病的認識。
1 臨床資料
例1 患者女性,33歲。因“咳嗽、咳痰1月余”于2016年6月1日入院。患者1個月前無明顯誘因出現咳嗽、咳痰,痰白量少,于2016年5月18日在當地醫院行肺部CT檢查示:兩肺多發病灶伴空洞形成。經抗感染治療后效果不佳轉來我院。患者既往有吸煙史(10包·年),近3個月來吸煙量有所增加,無粉塵接觸史。發現“子宮肌瘤”10余年。入院后體檢:體溫36.8℃,脈搏80次/min,呼吸20次/min,血壓108/62 mmHg。意識清楚,軀干、四肢無皮疹,淺表淋巴結未觸及腫大,雙肺語顫對稱,無增減,叩診清音,兩肺呼吸音清,未聞及干濕性啰音。心律齊,70次/min,無雜音。腹平軟,肝脾不大。雙下肢無浮腫。血生化檢測:肝、腎功能、電解質、血糖、血脂正常;動脈血氣分析:pH 7.39,PaO282mmHg,Pa-CO242mmHg,動脈血氧飽和度(SaO2)95%;血常規檢查:RBC 3.98×1012/L,Hb 91g/L,WBC 8.04×109/L,中性粒細胞71.1%,淋巴細胞14.1%,嗜酸性粒細胞3.7%;腫瘤標記物陰性;風濕全套:抗核抗體1:320,核型呈顆粒型,其余均陰性。腹部B超檢查示:肝、膽、脾、胰及雙腎未發現異常。顱骨、肋骨、骨盆、四肢長骨X線平片均未發現骨質破壞。副鼻竇X線平片未見急慢性鼻竇炎征像。肺功能檢測示:一秒用力呼氣容積與用力肺活量比值(FEV1/FVC)83.6%,FEV1占預計值90.2%,FVC占預計值72%,肺泡CO彌散功能正常。胸部增強CT檢查示:雙肺多發大小不等的類圓形結節樣高密度影,直徑4~11mm,病灶呈多樣性,可見實性結節、磨玻璃樣結節,部分融合,部分病灶內可見空洞,壁薄,中空,內無氣液平面。增強后實性結節強化不明顯(圖1a)。對比外院CT,肺部病灶增多,部分出現融合。氣管鏡下可見支氣管黏膜較蒼白,似有白色薄膜覆蓋,肺泡灌洗液細胞以淋巴細胞為主,占81%,灌洗液細菌培養、真菌培養陰性,經支氣管鏡肺活檢術(TBLB)提示未見惡性腫瘤細胞。對右肺下葉融合病灶行CT引導下經皮肺穿刺,病理提示:少量肺組織,肺泡腔內見少數組織細胞。為明確診斷,患者于2016年6月13日轉胸外科行胸腔鏡下左肺結節活檢術,病理報告示:左肺上葉結節內見上皮樣細胞聚集伴大量嗜酸性粒細胞浸潤,多灶性病變,小者最大徑約0.2cm,最大者體積1.0cm×0.8cm×0.6cm;免疫組化結果示:腫瘤細胞 S-100(+)、CD1a(+)、CD68(+)、LCA(+)、CK(pan)(-)、Ki-67(+)(陽性率<5%)(圖 2,見插頁)。明確診斷為LCH。囑患者嚴格戒煙。鑒于疾病進展快,給予強的松片10mg,3次/d,口服。間斷服用祛痰藥物鹽酸氨溴索。未行其他特殊治療。患者咳嗽癥狀消失,無其他不適主訴。1個月后復查胸部CT示雙肺結節性和囊性病變基本消失(圖1b)。
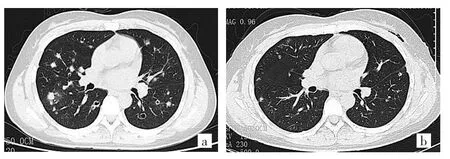
圖1 例1患者入院時及復查的胸部CT(a:雙肺多發大小不等的結節,部分病灶內可見壁薄空洞;b:病灶基本吸收)
例2 患者女性,69歲。因“軀干部紅色斑丘疹半年,頸部淋巴結腫大1個月”于2017年1月4日入院。患者半年前無誘因下逐步出現腋下、背部粟粒至硬幣大小紅斑、丘疹,伴瘙癢,有脫屑。曾就診皮膚科,考慮“銀屑病”,給予藥物外敷處理后未好轉,1個月前發現頸部淋巴結腫大,無畏寒、發熱、咳嗽、咳痰,無胸痛等不適,皮疹有加重趨勢,遂來我院就診。否認吸煙、嗜酒史。既往有“高血壓”病史10余年,口服纈沙坦80mg治療,血壓控制可。入院后體檢:體溫36.5℃,脈搏76次/min,呼吸19次/min,血壓135/90 mmHg。意識清楚,背部、腳踝處可見紅色斑疹,伴脫屑,雙側鎖骨及腋下、腹股溝可捫及腫大淋巴結,邊界清,無壓痛。兩肺呼吸音清,未聞及干濕性啰音。心律齊,心率76次/min,無雜音。腹平軟,肝脾不大。雙下肢無浮腫。血常規檢查:RBC 4.15×1012/L,Hb 12.7 g/L,WBC 3.49×109/L,中性粒細胞 55.1%,淋巴細胞30.9%,嗜酸粒細胞0.09%;血生化檢測:谷氨酰轉移酶 14 U/L,堿性磷酸酶80 U/L,AST 15U/L,ALT 9U/L;腫瘤標記物陰性;風濕全套:陰性。抗鏈球菌溶血素 O:<25U/ml;C 反應蛋白(CRP)、類風濕因子、甲狀腺功能均正常。患者腋下、腹股溝、頸部淋巴結B超檢查提示淋巴結增大。顱骨、肋骨、骨盆X線平片均未發現骨質破壞。骨髓活檢示:造血組織約占骨髓面積50%,粒紅比例正常,粒系各階段可見,未見幼稚粒細胞增多,紅系各階段可見,巨核細胞數量大致正常,各階段可見。胸部CT檢查示:左肺上葉多發小結節,炎性肉芽腫考慮;兩肺下葉局限間質性改變。患者于2017年1月12日行右側頸部淋巴結活檢術,病理報告示:淋巴組織增生,濾泡間大量組織細胞樣細胞浸潤,疑為組織細胞增生性病變,進一步免疫組化檢查結果示CD68(+)、CD1a(+)、S-100(+)、CD20生發中心(+)、CD79a生發中心(+)、Ki-67(+)(陽性率 20%)(圖 3,見插頁)。結果支持LCH。排除用藥禁忌后,于1月24日開始使用COP方案化療[環磷酰胺(CTX)0.8mg d1,長春地辛(VDS)4mg d1,地塞米松(DXM)15mg d1~5]4個療程,復查淋巴結明顯縮小,皮疹亦有所好轉,患者定期隨訪中。
2 討論
LCH是一種病因及發病機制均不明的少見疾病,發病率約為3/100~5/100萬,男性發病率高于女性[1]。目前研究認為LCH是由于朗格漢斯細胞的克隆性增殖而形成的腫瘤性疾病[2],主要特征是機體的不同器官組織發生肉芽腫性病變,可同時累及1個或多個器官組織[3]。LCH累及肺部可分為兩類:一類是肺部受累僅僅是全身多個器官病變之一,多見于小兒;另一類是單獨肺部受累,無其他器官病變,主要發生在成年人,如本文例1。單獨肺部受累的LCH非常罕見,國內外多為個案報道[4]。
超過90%的肺朗格漢斯細胞組織細胞增生癥(PLCH)發生于吸煙者,0~20歲是發病高峰。目前公認PLCH的發病與吸煙密切相關。煙霧可能是導致朗格漢斯細胞在肺內募集的始發因素[5]。吸煙者的肺中蛙皮素顯著增加,蛙皮素是神經內分泌細胞產生的一種神經肽類,對單核細胞有顯著的趨化作用,能夠促進上皮細胞和成纖維細胞的有絲分裂,并且刺激細胞因子的分泌[6]。煙草糖蛋白以及其他具有調節功能的糖肽(如粒細胞-巨噬細胞集落刺激因子、IL-2、TNF)也可能對發病起到一定的作用[6]。本文例1患者為年輕女性,且有長期吸煙史,與上述一致。
除肺以外,LCH可發生在骨骼系統、皮膚、淋巴結、肝臟、脾臟、生殖器、腎臟及胸腺等。如本文例2同時累及全身淋巴結及皮膚。許霞等[7]對160例確診為LCH患者的臨床特征分析提示淋巴結受累者占8.1%,皮膚損害占3.1%;但朱里等[8]總結了1994-2007年國內期刊上發表的個案報道及回顧性分析,共納入918例患者,其中有皮膚損害510例(55.5%),文獻報道之間差異較大。目前皮膚病變型LCH在成人中的真正發病率尚不清楚,可能有些皮膚病損較輕或為早期而難以識別,因而被漏診或誤診。尚需要大樣本多中心臨床研究進一步明確。
由于成人LCH患者的臨床特征無特異性,并且該病多發生于兒童,很容易被臨床醫師誤診、誤治,誤診率為7.5%~59.1%[9-10]。本文中2例患者均存在不同程度誤診,例1曾被誤診為肺惡性腫瘤、肺真菌感染;例2被誤診為淋巴瘤、銀屑病。分析誤診原因可能為本病成人中發病率低,導致臨床醫師對此病診斷經驗不足;另一原因是此病臨床表現復雜多樣,且無特征性,尤其是早期病變,臨床癥狀較輕,更加難以識別。因此,在臨床實踐中,我們應開拓思路,高度重視與LCH的鑒別。LCH確診依賴于病理,通常在光鏡下可見朗格漢斯細胞,有特異性核溝,免疫組化檢查膜表面CD1a抗原、CD68抗原、S-100蛋白均應陽性[11],本文患者淋巴結及肺結節切除腫塊免疫組化檢查符合上述結果。
LCH的治療方法包括手術治療、放療、化療,或幾種方法的聯合治療。對于單獨肺部受累患者,戒煙是目前最常采用的治療方法,戒煙可以穩定病情,減緩病變進展,甚至有報道發現戒煙可使患者主觀癥狀和CT表現完全消失[12]。但戒煙對患者長期生存率和病死率的影響有待進一步闡明。對于臨床癥狀較重或進展較快的患者可使用糖皮質激素治療。初始劑量為潑尼松0.5~lmg/(kg·d),之后逐漸減量,連續服用 6~12 個月[13]。例 1 患者肺部CT進展快,結節病灶出現融合,我們在指導其戒煙基礎上,給予糖皮質激素口服治療后效果顯著,肺部影像學表現迅速好轉,與上述相符。對于慢性復發性、頑固性、進展性多系統受累患者,目前尚無統一治療方案。Gadner等[14]研究表明,長春新堿聯合甲潑尼龍方案可作為LCH多系統疾病者的一線治療方案;Girschikofsky等[15]報道阿糖胞苷、依托泊苷、克拉屈濱等也可作為LCH的化療藥物;此外,最近有研究發現BRAFV600E基因突變可見于部分LCH患者,將針對該突變的靶向藥物威羅菲尼用于伴BRAFV600E基突變陽性的LCH患者,可取得較好的療效[16-17],為該病提供了一種新的治療方法。本文例2應用COP方案4個療程后獲得了較好的療效,也進一步證實了上述結論。
LCH患者一般預后良好,在治療后病情趨于穩定甚至治愈,但部分患者可出現尿崩癥、生長激素不足、長期肝臟和肺功能異常、不伴有肝功能異常的膽管炎、脊柱或肢體的畸形等并發癥。本文2例患者尚未見上述并發癥,隨訪至今,目前情況良好。
[1]Zinn DJ,Chakraborty R,Allen CE.Langerhans Cell Histiocytosis:Emerging Insights and Clinical Implications[J].Oncology(Williston Park),2016,30(2):122-132,139.
[2]Suri HS,Yi ES,Nowakowski GS,et al.Pulmonary langerhans cell histiocytosis[J].Orphanet J Rare Dis,2012,7:16.
[3]徐葉惠,劉淑萍,周曄敏.1例中樞神經系統朗格漢斯組織細胞增生癥患者的護理[J].浙江醫學,2015,37(3):259-261.
[4]Seely JM,Salahudeen S,Sr.,Cadaval-Goncalves AT,et al.Pulmonary Langerhans cell histiocytosis:a comparative study of computed tomography in children and adults[J].J Thorac Imaging,2012,27(1):65-70.
[5]王全,夏雯,趙德育.單獨肺受累的兒童肺郎格罕細胞組織細胞增生癥一例報道并文獻分析[J].中華兒科雜志,2012,50(2):146-150.
[6]Vassallo R,Ryu JH,Colby TV,et al.Pulmonary Langerhans'-cell histiocytosis[J].N EnglJ Med,2000,342(26):1969-1978.
[7]許霞,聶秀,熊文,等.160例成人朗格漢斯細胞組織細胞增生癥患者臨床特征分析[J].中華血液學雜志,2015,36(2):135-139.
[8]朱里,黃長征,陳思遠,等.朗格漢斯細胞組織細胞增生癥皮膚損害臨床分析[J].中國麻風皮膚病雜志,2008,24(10):787-791.
[9]葛榮,殷憲剛,劉創峰,等.成人朗格漢斯細胞組織細胞增生癥的臨床病理學分析[J].中華醫學雜志,2012,92(42):2995-2997.
[10]韓瀟,周道斌,焦力,等.成人郎格罕斯細胞組織細胞增生癥40例臨床表現分析[J].中華血液學雜志,2010,31(10):699-701.
[11]Harmon CM,Brown N.Langerhans CellHistiocytosis:A Clinicopathologic Review and Molecular Pathogenetic Update[J].Arch P-atholLab Med,2015,139(10):1211-1214.
[12]Elia D,Torre O,Cassandro R,et al.Pulmonary Langerhans cell histiocytosis:a comprehensive analysis of 40 patients and literature review[J].Eur J Intern Med,2015,26(5):351-356.
[13]Shen J,Feng S.Bone Langerhans cell histiocytosis with pulmonary involvement in an adult non-smoker:Acase report and brief review of the literature[J].MolClin Oncol,2017,6(1):67-70.
[14]Gadner H,Minkov M,Grois N,et al.Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis[J].Blood,2013,121(25):5006-5014.
[15]Girschikofsky M,Arico M,Castillo D,et al.Management of adult patients with Langerhans cell histiocytosis:recommendations from an expert panelon behalf of Euro-Histio-Net[J].Orphanet J Rare Dis,2013,8:72.
[16]Roden AC,Yi ES.Pulmonary Langerhans Cell Histiocytosis:An Update From the Pathologists'Perspective[J].Arch PatholLab M-ed,2016,140(3):230-240.
[17]Satoh T,Smith A,Sarde A,et al.B-RAF mutant alleles associated with Langerhans cell histiocytosis,a granulomatous pediatric disease[J].PLoS One,2012,7(4):e33891.
