PARP表達量降低對氧化應激損傷人心肌細胞凋亡的抑制作用及機制
2018-03-19 13:31:33閆振富
山東醫藥 2018年1期
閆振富
(鄭州大學附屬鄭州中心醫院,鄭州450000)
冠狀動脈粥樣硬化性心臟病、慢性心力衰竭、高血壓等心血管疾病病死率和發病率較高,嚴重危害人類的健康。氧化應激是誘導產生心血管疾病的重要原因之一[1]。機體在正常的狀態下,體內氧自由基和活性氧(ROS)的產生及消除維持著動態平衡。而當機體受到有害刺激時,氧自由基、ROS等氧化產物的生成遠大于機體消除能力,從而損傷組織和細胞的抗氧化系統,導致細胞氧化應激損傷。因此研究氧化應激引起的心肌細胞凋亡可以有效改善心血管疾病的治療。研究證實,ROS可抑制心肌細胞增殖、誘導其凋亡或壞死,加重心血管病情[2]。過氧化氫(H2O2)是一種重要的ROS,氧化能力較強,常用于建造氧化應激損傷模型。聚腺苷二磷酸核糖聚合酶(PARP)是一種多功能酶,主要作用是促進DNA修復、復制、轉錄調控和細胞分化等。研究表明PARP與心肌損傷或梗死等病理生理過程密切相關[3]。2014年6月~2016年10月本研究將siRNA-PARP轉染入H2O2損傷的人心肌細胞H9C2中,以探討PARP表達量降低對氧化應激損傷心肌細胞增殖、凋亡的作用及機制。
1 材料與方法
1.1 主要材料 人心肌細胞H9C2、Eagle's Minimum Essential Medium(EMEM)培養基均購自美國American Type Culture Collection公司,胎牛血清(FBS)、Lipofectamine 2000轉染試劑盒均購自美國Invitrogen公司,細胞計數試劑盒(CCK-8)購自武漢默沙克生物科技有限公司,AnnexinV FITC細胞凋亡檢測試劑盒購自美國BD公司,BCA蛋白定量試劑盒購自上海碧云天生物技術研究所,蛋白激酶B(AKT)一抗、磷酸化AKT(pAKT)一抗、磷脂酰肌醇-3激酶(PI3K)一抗、磷酸化PI3K(pPI3K)一抗、甘油醛-3-磷酸脫氫酶(GAPDH)一抗、辣根過氧化物酶標記羊抗兔二抗購自美國Santa Cruz公司等。
1.2 細胞培養及模型建立 將人心肌細胞H9C2培養于EMEM培養基中,加入10% FBS、100 U/mL青霉素、100 mg/mL鏈霉素,保證培養環境無細菌污染,置于5% CO2、溫度37 ℃和濕度95%的細胞培養箱中培養。每天在倒置顯微鏡下觀察細胞的生長狀態,使用0、200 μmol/L H2O2混合氣體培養細胞48 h,建造氧化應激模型。
1.3 細胞轉染及效果檢測 按照Lipofectamine 2000的說明書進行脂質體轉染。取對數期細胞作為實驗對象,分為4組即對照組1、對照組2、陰性組、干擾組。轉染前1 d,調整細胞濃度,每孔1×105個細胞和2 mL培養基接種于6孔板中,置于5% CO2、37 ℃的細胞培養箱中培養24 h,待細胞融合度達到70%~80%左右進行轉染,加入5 μL脂質體Lipofectamine 2000和100 μL無血清培養基,陰性組加入siRNA-NC,干擾組加入siRNA-PARP,對照組2、陰性組、干擾組細胞均為200 μmol/L H2O2刺激的細胞,對照組1為0 μmol/L H2O2刺激的細胞,室溫孵育20 min,放置培養箱中連續培養5 h后更換新鮮培養基繼續培養,24 h后Western blotting檢測轉染效果。
1.4 細胞增殖率檢測 采用CCK-8。取各組轉染后24 h細胞進行實驗,以每孔1×105個細胞接種于96孔板上,置于5% CO2、37 ℃的細胞培養箱中培養24 h。每組5個復孔,加入10 μL/孔CCK-8溶液,孵育2 h后置于酶標儀上測定450 nm波長下的吸光值。實驗重復3次。細胞增殖率=(對照組平均吸光值/實驗組平均吸光值)×100%。
1.5 細胞凋亡率檢測 采用流式細胞術。胰酶消化各組對數期細胞,1×Binding Buffer緩沖液將細胞密度調整為1×105個/mL,接種于6孔板中,待細胞融合度為70%~80%時,加入10 μL膜聯蛋白V-FITC和10 μL碘化丙啶,振蕩混勻,置于流式細胞儀上檢測細胞的凋亡率,每組5個復孔取平均數,實驗重復3次。
1.6 PI3K、pPI3K、AKT、pAKT蛋白表達量檢測 采用Western blotting法。收集各組細胞加入500 μL RIPA裂解液,充分混勻,置于冰上反應30 min,4 ℃ 12 000 r/m離心10 min,取上清即為提取的總蛋白。根據BCA蛋白定量說明書檢測蛋白濃度。將提取的蛋白放入沸水中變性5 min。取50 μg蛋白樣品,使用10%的分離膠和5%的濃縮膠進行SDS-PAGE凝膠電泳至溴酚藍到達凝膠底部,然后進行轉膜70 min,在5%脫脂奶粉中室溫封閉2 h,剪取含有目的條帶和內參GAPDH的條帶,分別加入一抗,4 ℃孵育過夜,TBST液漂洗,10 min×3次,加入二抗,37 ℃反應2 h,TBST液漂洗,10 min×3次,加入ECL化學發光液,避光反應10 min,分析蛋白質的灰度值,以目的條帶灰度值與內參GAPDH灰度值的比值為各目的蛋白的相對表達量。
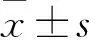
2 結果
2.1 各組細胞轉染效果比較 對照組1、對照組2、陰性組、干擾組心肌細胞中PARP蛋白的表達量分別為0.213±0.026、0.585±0.064、0.561±0.062、0.383±0.032。與對照組1相比,對照組2、陰性組和干擾組細胞中PARP蛋白的表達量增加(P均<0.05); 與對照組2相比,陰性組心肌細胞中PARP蛋白的表達量差異無統計學意義(P>0.05),但干擾組PARP蛋白的表達量降低(P<0.05)。
2.2 各組細胞增殖率比較 對照組1、對照組2、陰性組、干擾組心肌細胞的增殖率分別為100.000%±0.062%、62.143%±5.967%、60.148%±5.814%、83.452%±8.785%。與對照組1相比,對照組2、陰性組、干擾組心肌細胞的增殖率明顯降低(P均<0.05);與對照組2相比,陰性組心肌細胞的增殖率差異無統計學意義(P>0.05),但干擾組心肌細胞的增殖率升高(P<0.05)。
2.3 各組細胞凋亡率比較 對照組1、對照組2、陰性組、干擾組心肌細胞的凋亡率分別為8.421%±0.830%、27.573%±2.269%、28.205%±2.597%、15.648%±1.521%。與對照組1相比,對照組2、陰性組、干擾組心肌細胞的凋亡率明顯升高(P均<0.05);與對照組2相比,陰性組心肌細胞的凋亡率差異無統計學意義(P>0.05),但干擾組心肌細胞的凋亡率降低(P<0.05)。
2.4 各組PI3K、pPI3K、AKT、pAKT蛋白表達量比較 對照組1、對照組2、陰性組、干擾組心肌細胞中PI3K蛋白的表達量為0.785±0.082、0.386±0.031、0.362±0.035、0.512±0.053,pPI3K的表達量為0.712±0.075、0.322±0.034、0.331±0.026、0.523±0.049,AKT的表達量為0.878±0.072、0.461±0.044、0.442±0.042、0.626±0.057,pAKT的表達量為0.850±0.079、0.446±0.042、0.424±0.041、0.611±0.058。與對照組1相比,對照組2、陰性組、干擾組心肌細胞中PI3K、pPI3K、AKT、pAKT的表達量降低(P均<0.05);與對照組2相比,陰性組心肌細胞中PI3K、pPI3K、AKT、pAKT表達量差異無統計學意義(P均>0.05)、干擾組心肌細胞中PI3K、pPI3K、AKT、pAKT的表達量增加(P均<0.05)。
3 討論
RNA干擾技術主要通過雙鏈RNA 引起同源轉錄因子降解,特異性阻礙其翻譯生成蛋白質分子,從而沉默基因的現象。由于RNA干擾技術具有特異性、高效性等特點,廣泛應用于疾病的基因治療、新藥開發、基因功能、神經系統等的研究中,為癌癥的治療及遺傳病的研究提供了新的思路和方法。RNA干擾技術首次應用于線蟲體內,結果發現注入正義鏈和反義鏈的混合物比單獨注入單鏈RNA引起基因沉默的效果要強很多,且可沉默子一代同源基因。本研究通過siRNA靶向沉默H9C2心肌細胞PARP基因的表達,結果顯示,PARP基因的表達量顯著下降。
PARP屬于真核細胞中DNA修復酶之一,與DNA的損傷和修復有關,主要通過與細胞凋亡相關蛋白的結合調控細胞的凋亡過程。當細胞發生凋亡時,PARP不能與DNA片段結合,影響其損傷修復能力,導致DNA裂解,誘導細胞凋亡[4]。研究顯示,PARP在心肌損傷、慢性心力衰竭等心臟疾病的生理過程中發揮重要作用[5]。PARP在心肌損傷后被激活,存在于心臟疾病的整個病理生理改變過程中[6]。當細胞凋亡時,PARP被過度激活,耗盡細胞內的能量物質從而誘導細胞凋亡、損害機體[7]。ROS的過度積累導致細胞發生氧化應激,是導致細胞凋亡、損傷的主要原因之一,在心血管疾病的發生、發展過程中起著重要作用。H2O2屬于ROS的一種,常被用來建造體外心肌細胞、神經元等氧化應激損傷模型。研究表明,以200 μmol/L H2O2可以降低心肌細胞50%的活力[8,9]。因此本實驗通過200 μmol/L H2O2刺激心肌細胞H9C2建造氧化應激損傷模型,觀察PARP的表達量降低對心肌細胞損傷的保護作用。流式細胞術實驗結果表明H2O2可促進細胞凋亡,低表達的PARP可抑制氧化應激引起的心肌細胞凋亡、促進其增殖。
細胞的凋亡過程受多條信號通路的調控。PI3K/AKT信號通路是一條絡氨酸激酶級聯信號傳導通路,主要通過影響下游靶基因的表達調控細胞的生長、分化、侵襲和促進血管生成、改善器官等[10]。近年來的研究表明機體細胞發生氧化應激時,PI3K/AKT信號通路可誘導心臟、大腦等細胞發生凋亡[11]。AKT是PI3K重要的下游靶基因,AKT和磷酸肌醇依賴性蛋白激酶-1可與PI3K催化的3,4二磷酸磷脂酰肌醇結合,促使AKT磷酸化,調控下游凋亡蛋白的表達,參與細胞增殖、分化等生理過程。研究報道,pAKT可促使多種細胞凋亡相關基因的表達下降或失活,具有抑制細胞凋亡的作用,同時也可以減少氧化應激誘導的細胞損傷。本研究中,H2O2刺激后導致心肌細胞增殖率降低、凋亡率增加,發生氧化應激損傷;Western blotting檢測發現氧化應激損傷的細胞內PI3K、pPI3K、AKT和pAKT的表達量顯著下降,促進細胞凋亡的作用增強。沉默PARP表達緩解氧化應激引起PI3K、pPI3K、AKT和pAKT的表達量降低,提示PARP可能是通過PI3K/AKT途徑,發揮改善H2O2抑制PI3K/AKT信號通路的作用。
綜上所述,H2O2刺激可使心肌細胞發生氧化應激損傷,細胞的凋亡率增加、增殖率降低,但RNA干擾PARP表達能通過調控PI3K/AKT信號通路,抑制H2O2誘導的H9C2細胞凋亡,促進其增殖,有效保護心肌細胞。
[1] Quintana-Villamandos B, Delgado-Baeza E. Does the ADMA/DDAH/NO pathway modulate early regression of left ventricular hypertrophy with esmolol[J]. Med Hypotheses, 2016,87(2):44-47.
[2] 王全偉,凡文博,王智昊,等.氧化應激與心血管疾病關系的研究進展[J].中國老年學雜志,2014,34(1):270-273.
[3] Han J, Kim B, Shin JY, et al. Iron oxide nanoparticle-mediated development of cellular gap junction crosstalk to improve mesenchymal stem cells'therapeutic efficacy for myocardial infarction[J]. ACS Nano, 2015,9(3):2805-2819.
[4] 凌曉璇,溫橋生,杜諭君,等.多聚聚合酶-1在氫醌誘導的DNA損傷應答中的作用及其機制[J].職業與健康,2015,31(14):1906-1909.
[5] Bacalini MG, Tavolaro S, Peragine N, et al. A subset of chronic lymphocytic leukemia patients display reduced levels of PARP1 expression coupled with a defective irradiation-induced apoptosis[J]. Exp Hematol, 2012,40(3):197-206.
[6] Ger D, Szoleczky P, Chatzianastasiou A, et al. Modulation of poly (ADP-ribose) polymerase-1 (PARP-1)-mediated oxidative cell injury by ring finger protein 146 (RNF146) in cardiac myocytes[J]. Mol Med, 2014,20(1):313-328.
[7] Erikson JM, Valente AJ, Mummidi S, et al. Targeting TRAF3IP2 by genetic and interventional approaches inhibits ischemia/reperfusion-induced myocardial injury and adverse remodeling[J]. J Biol Chem, 2017,292(6):2345-2358.
[8] 鐘源,孫善全.番茄紅素對心肌細胞氧化應激損傷的保護作用[J].重慶醫學,2015,44(9):1157-1161.
[9] Wang W, Wang L, Yang H, et al. Protective effects of yindanxinnaotong capsule in a rat model of myocardial ischemia/reperfusion injury[J]. J Tradit Chin Med, 2014,34(6):699-709.
[10] Shohei M, Yasuaki F, Haruka T, et al. Mechanism of the inhibition of leukemia cell growth and induction of apoptosis through the activation of ATR and PTEN by the topoisomerase inhibitor 3EZ, 20Ac-ingenol[J]. Leuk Res, 2015,39(9):927-932.
[11] 韓貴賓,張壽,孫薇薇,等.尼古丁抑制MIA誘導的骨關節炎軟骨細胞凋亡[J].中國比較醫學雜志,2016,26(3):40-45.
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
世界科學技術-中醫藥現代化(2020年2期)2020-07-25 02:05:56
海峽科技與產業(2016年3期)2016-05-17 04:32:12
西南軍醫(2016年6期)2016-01-23 02:21:19
新疆醫科大學學報(2015年10期)2015-12-26 12:33:30
癌變·畸變·突變(2015年3期)2015-02-27 06:15:09
