海水吸入性急性呼吸窘迫綜合征發病機制的研究進展
2018-03-12 09:10:48謝永宏金發光
中華肺部疾病雜志(電子版) 2018年1期
謝永宏 金發光
海水淹溺是一個重要但又經常被忽視的公共衛生問題。20世紀90年代末世界銀行和WTO頒布的第一個全球疾病負擔(global burden of disease, GBD)就指出海水淹溺是最常見的死亡原因之一。據保守估計,每年全球約有50萬人死于海水淹溺,非致死性溺水發生率比致死性溺水高出2倍以上,而且海水淹溺導致的急性肺損傷往往比淡水淹溺更為嚴重[1-2]。2008年發布的《世界兒童傷害預防報告》也指出非致命性溺水對終身健康和經濟造成的影響超過其他任何傷害。盡管淹溺發生多、致死致殘率高,但國際上對海水淹溺性肺損傷的研究并不多,國內外也未見關于海水淹溺性肺損傷發病機制的系統研究。Orlowski等[3-4]在上世紀八十年代末進行了部分海水淹溺性肺損傷相關基礎研究,明確吸入淡水或海水后動物出現呼吸困難,頻率加快,兩肺布滿濕啰音,氣管內溢出泡沫狀液體。PaO2、SaO2、血液pH值、實際碳酸氫鹽(AB)顯著降低,表明出現了進行性低氧血癥和代謝性酸中毒,結合病理改變證明出現了肺水腫[5]。然而,海水吸入性急性呼吸窘迫綜合征(seawater-aspiration acute respiratory distress syndrome, SW-ARDS)的救治和基礎研究用于SW-ARDS的救治在國際上卻比較少見。
近年來,隨著我國海洋活動增加,SW-ARDS的救治和基礎研究又逐漸引起國內學者的重視,并在SW-ARDS發病機制方面取得一定的成果。明確了炎癥、氧化應激、細胞凋亡和細胞間通訊障礙等機制參與了SW-ARDS的發生和發展;逐步深入研究發現,除了這些多種肺損傷中均存在的病理機制以外,海水自身高滲特點和吸入海水后所引起的機體缺氧可能也參與了SW-ARDS的發生與發展[6-9]。如果在SW-ARDS發生發展過程中,早期能夠準確預警,中期進行有效治療,晚期進行積極救治,就可以明顯改善其預后。但遺憾的是,在此之前,其具體發病機制一直不夠明確,發生發展的關鍵環節也不清楚,救治方法始終不理想。
目前,人們已經逐漸認識到炎癥反應是SW-ARDS的主要發病機制,參與炎癥反應的細胞主要有多形核白細胞、巨噬細胞、血管內皮細胞、肺泡上皮細胞等。活化炎癥細胞釋放大量炎性介質,如彈性蛋白酶、組織蛋白酶、膠原酶、明膠酶、腫瘤壞死因子(tumor necrosis factor-α, TNF-α)、白細胞介素-1β(interleukin-1β, IL-1β)、IL-9、IL-8和血小板活化因子等,這些炎性介質作用于肺毛細血管內皮細胞和肺泡上皮細胞等誘發肺損傷[10-13]。既然全身和局部的炎癥反應是ARDS發生和發展的重要機制,理論上糖皮質激素應當具有良好的治療效果。然而,大量試圖應用糖皮質激素控制炎癥反應,預防和治療ARDS的研究結果表明,糖皮質激素既不能預防ARDS的發生、甚至對早期肺損傷也沒有治療作用[14]。究其原因,不同的損傷因素所導致的ARDS既有相似臨床特征,又有不同表現形式,這可能預示其發病機制既具有共同的作用環節又有特異性的信號途徑,如果能夠明確SW-ARDS發生發展的關鍵分子靶點并篩選出相應的抗炎藥物將極有可能最終破解這一難題。現就SW-ARDS的發病機制研究的新進展做一全面綜述,以便提高人們對SW-ARDS的深入認識。
一、SW-ARDS的動物模型制作
第四軍醫大學唐都醫院在國內成功構建了大鼠SW-ARDS的動物模型,為后續研究奠定了基礎。其制作方法為:用3%戊巴比妥鈉(1.5 ml/kg)腹腔內注射的方式麻醉大鼠,仰臥固定并保持頭部抬高45度,將大鼠舌頭拉出,避免舌后墜窒息。右側頸總動脈插管以備取血測量PaO2和PaCO2;采取甲狀軟骨處頸部正中切口,鈍性分離暴露氣管,取1 ml注射器刺入氣管,以4 ml/kg海水緩慢注入氣管內,在4 min內完成。大鼠迅速出現耳鼻發紺、呼吸急促,自口、鼻腔涌出粉紅色泡沫樣液體,雙肺聽診布滿濕性啰音,動脈血氣示PaO2<50 mmHg,SaO2<45%左右,PaO2/FiO2(氧合指數)<150 mmHg提示海水淹溺模型建立成功[7]。
二、SW-ARDS發病機制
通過成功構建大鼠SW-ARDS動物模型的研究發現,當ARDS形成時均存在著缺氧、炎癥反應、肺組織細胞凋亡等基本病理生理變化,并通過對炎癥反應、高滲反應、細胞凋亡等多個信號通路的研究中明確了SW-ARDS發病機制。
1. 高滲啟動、HIF-1α介導、PI3K/AKT信號途徑激活是SW-ARDS發生發展的關鍵分子機制: 在研究過程中,通過建立不同滲透壓梯度動物模型,發現高滲是海水淹溺性肺損傷最主要的啟動因素,并誘導了HIF-1α高表達,引起肺組織缺氧。研究證實HIF-1α可激活PI3K/AKT信號途徑,并引起下游炎癥因子過度反應,導致肺組織原發性損傷。得出了以HIF-1α為中心的關于SW-ARDS肺損傷的理論,見圖1。
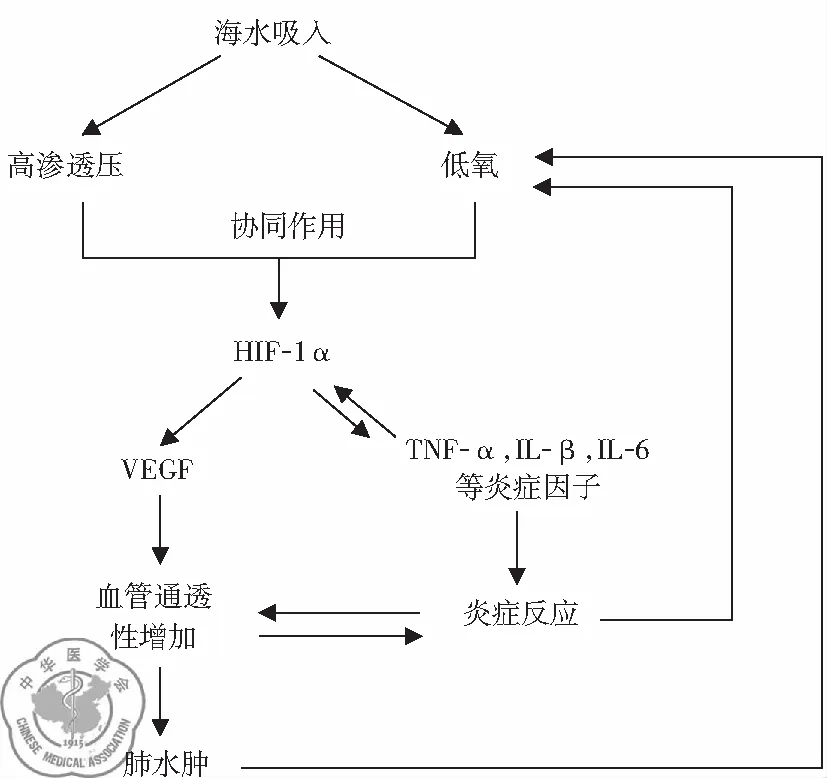
圖1 HIF-1α在海水淹溺性肺損傷機制中的作用
海水淹溺導致肺部高滲、缺氧環境,高滲與缺氧協同作用更明顯地促進肺組織HIF-1α的表達,增加的HIF-1α分別通過“HIF-1α-VEGF-肺血管通透性-肺水腫”和“HIF-1α-前炎癥因子(TNF-α、IL-1β、IL-6)-肺組織炎癥反應”兩條路徑加重肺血管通透性、肺水腫、肺組織炎癥反應,進而使肺組織缺氧更加嚴重,形成一個以HIF-1α為中心的肺損傷逐漸加重的惡性循環[15]。
2. 高滲啟動HIF-1α介導、Semaphorin 7A信號途徑參與SW-ARDS的機制形成: 通過構建SW-ARDS大鼠模型的研究表明,海水吸入導致肺組織處于高滲缺氧的環境,該環境促進肺組織HIF-1 α的表達,HIF-1 α表達的增高又引起了血管內皮細胞SEMA7A表達的上調,增加的SEMA7A通過與內皮Plexin C1受體作用,引起血管內皮生長因子(vascular endothelial growth factor, VEGF)表達上調,最終導致肺血管通透性增加。另外上調的SEMA7A還通過與滲出的炎癥細胞β1 integrin受體相互作用,激活p38MAPK以及NF-κB通路,最終加強肺組織炎癥反應。而肺血管通透性增加以及炎癥反應又進一步使肺組織缺氧更加嚴重,形成了一個以SEMA7A為中心的肺損傷逐漸加重的惡性循環[16],見圖2。
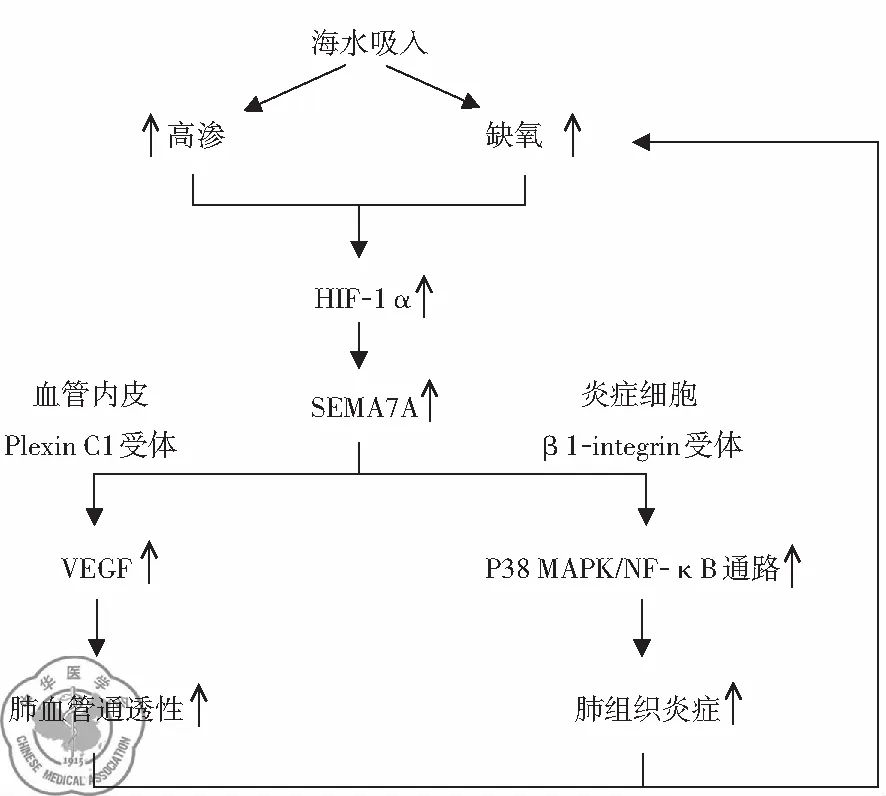
圖2 “HIF-1α-SEMA7A途徑”在SW-ARDS中的作用機制示意圖
3. ANGⅡ/ANG1-7內質網應激反應并誘導肺組織細胞凋亡: 通過動物實驗,及對細胞和多種分子生物學實驗研究,檢測SW-ARDS時肺組織中ANGⅡ/Ang1-7體系的表達,并進一步明確了ANGⅡ/Ang1-7體系在海水刺激下內質網應激誘導凋亡發生的機制。首次研究了Ang1-7在海水刺激后內質網應激過程中的變化,并驗證了Ang1-7對內質網應激誘導凋亡的保護作用和機制。最終,我們提出了ANGⅡ/Ang1-7體系參與介導海水淹溺性肺損傷時內質網應激反應誘導凋亡的作用機制理論[10],見圖3。

圖3 ANGII/ Ang1-7體系在海水吸入型肺損傷中內質網應激反應誘導凋亡的作用機制
4. JNK-mtDNA-中性粒細胞胞外損傷途徑參與SW-ARDS的形成: 研究顯示海水可以刺激肺臟的組織細胞,擾亂細胞及線粒體的正常結構及功能,導致產生大量的mtDNA并釋放到肺泡腔及肺細胞間隙中。釋放的這些mtDNA可以損傷肺臟的細胞、破壞血氣屏障,也可以憑借自身的免疫原性激活機體的免疫系統,引起中性粒細胞的黏附、滲出以及炎性因子的釋放。mtDNA所引起的過度激活的免疫系統,尤其是中性粒細胞的激活,進一步加重了肺損傷的程度。實驗證實mtDNA以及NETs的形成及產生損傷作用主要依賴于ROS,在肺組織細胞處于氧化應激狀態時,更易受到mtDNA及NETs的破壞。而JNK-線粒體通路的激活可以引起線粒體功能紊亂,造成ROS在細胞內的積聚。通過特異性多肽Tat-SabKIM1可以抑制海水刺激時肺臟細胞的JNK向線粒體轉移,繼而維持線粒體的穩定,阻斷JNK-線粒體通路激活,起到抑制細胞自噬及凋亡、減輕肺損傷的作用,見圖4。

圖4 海水吸入通過“JNK-mtDNA-NETs”途徑介導肺組織損傷示意圖
5. TonEBP/NFAT5信號通路介導肺組織損傷: 研究顯示TonEBP/NFAT5信號通路參與SW-ARDS的發生發展,TonEBP/NFAT5的表達及活化受到p38 MAPK信號通路的調節。配方海水、高滲NaCl溶液和LPS刺激都能夠引起細胞中p38 MAPK信號通路的異常活化;配方海水和高滲NaCl溶液對TonEBP/NFAT5及其下游分子表達的影響更為明顯,但同比例的配方海水對TonEBP/NFAT5的影響強于高滲NaCl溶液;這說明以TonEBP/NFAT5為核心的信號通路是SW-ARDS發病機制中的獨特環節。TonEBP/NFAT5通過調節Na+-K+-ATP酶的活性和表達參與調控SW-ARDS時肺水腫的發生[17],見圖5。
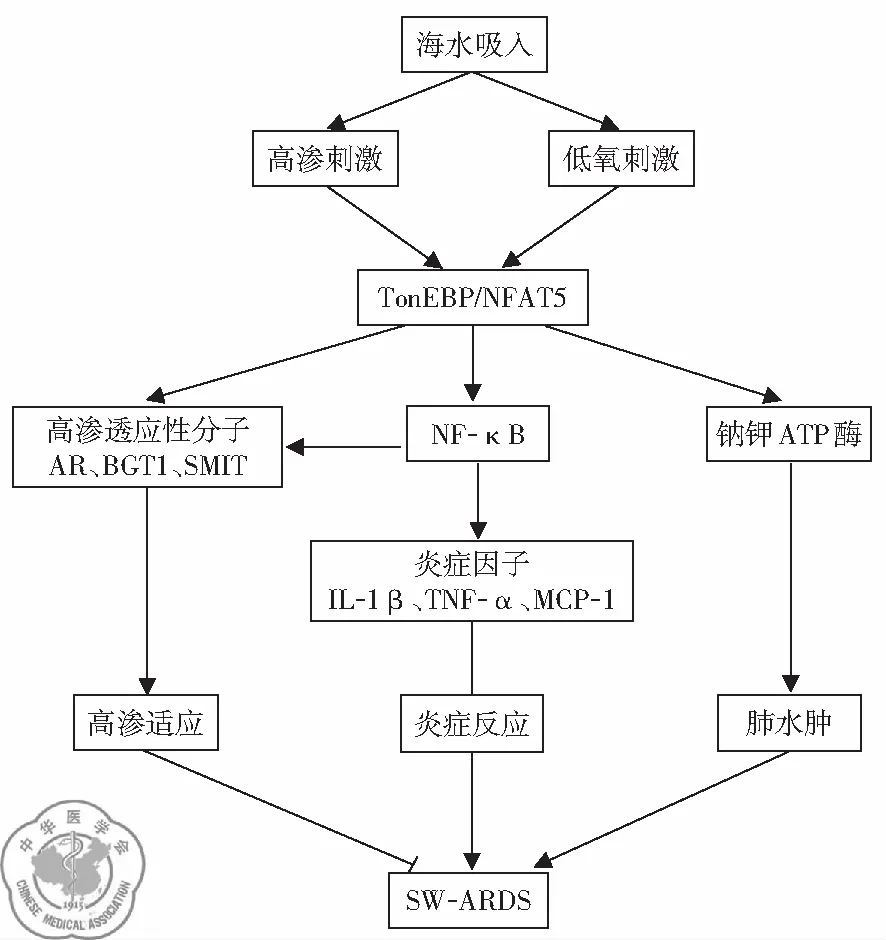
圖5 TonEBP/NFAT5信號通路介導SW-ARDS機制示意圖
6. p38 MAPK-p53途徑”和“Fas/FasL途徑”介導肺組織細胞凋亡: 新近研究表明野生型p53及其下游目的基因通過內源性途徑調節了海水淹溺性肺損傷時肺泡上皮細胞的凋亡;Fas/FasL介導的細胞凋亡途徑參與海水刺激時肺組織細胞的凋亡。此外,p38 MAPK作為p53的上游因子,通過磷酸化p53,部分地介導了海水吸入誘導的細胞凋亡調節[18],見圖6。
綜上所述,通過構建穩定的SW-ARDS大鼠模型,觀察海水刺激后肺組織的損傷變化,發現SW-ARDS發生時存在缺氧、炎癥反應、肺水腫、肺組織細胞凋亡4種基本病理生理變化。以吸入海水導致機體缺氧和肺組織局部高滲刺激為切入點,系統研究SW-ARDS的發病機制,先后明確了海水刺激通過“TonEBP/NFAT5-HIF-1α/NF-κB信號通路”和“JNK—線粒體DNA(mtDNA)—中性粒細胞胞外殺菌網絡(NETs)”介導肺部炎癥反應和肺水腫的機制,海水吸入導致ANGII/ANG1-7體系、“p38 MAPK-p53途徑”和“Fas/FasL途徑”異常表達引起肺組織細胞凋亡的機制。總體而言,肺水腫、肺部炎癥反應和肺組織細胞凋亡三個方面共同引起海水吸入導致SW-ARDS的發生。因而,在海水淹溺救治中應當從這些途徑入手,尋找診斷和治療SW-ARDS的新方法。
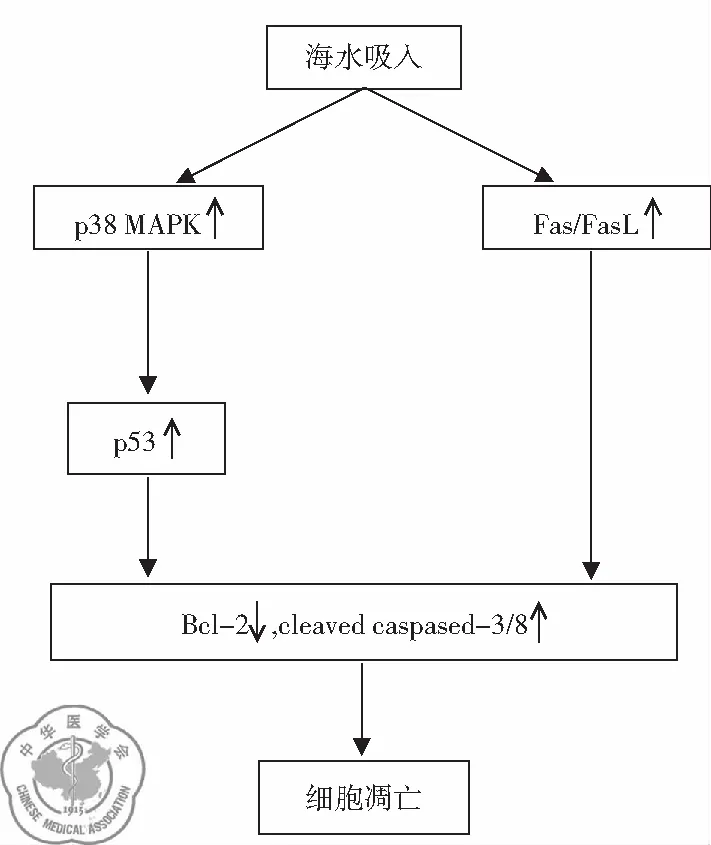
圖6 海水吸入導致肺組織細胞凋亡示意圖
1 李鵬程, 李聰聰, 魯曦, 等. 海水干預下人肺泡上皮細胞活性氧及內質網應激水平的變化[J/CD]. 中華肺部疾病雜志(電子版), 2016, 9(5): 489-493.
2 李聰聰, 薄麗艷, 劉慶晴, 等. 蛋白激酶SPAK在海水淹溺性肺損傷中的表達和作用[J/CD]. 中華肺部疾病雜志(電子版), 2014, 7(1): 19-22.
3 Orlowski JP. Drowning, near-drowning, and ice-water submersions[J]. Pediatr Clin North Am, 1987, 34(1): 75-92.
4 Orlowski JP. Drowning, near-drowning, and ice-water drowning[J]. JAMA, 1988, 260(3): 390-391.
5 Gu MN, Xiao JF, Huang YR, et al. Study of direct lung injury by seawater in canine models[J]. Di Yi Jun Yi Da Xue Xue Bao, 2003, 23(3): 201-205.
6 Ji MH., Tong JH., Tan YH., et al. Erythropoietin pretreatment attenuates seawater aspiration-induced acute lung injury in rats[J]. Inflammation, 2016, 39(1): 447-456.
7 Ma L, Chen X, Wang R, et al. 3,5,4′-Tri-O-acetylresveratrol decreases seawater inhalation-induced acute lung injury by interfering with the NF-kappaB and i-NOS pathways[J]. Int J Mol Med, 2016, 37(1): 165-172.
8 Diao M., Zhang S., Wu L., et al. Hydrogen Gas Inhalation Attenuates Seawater Instillation-Induced Acute Lung Injury via the Nrf2 Pathway in Rabbits[J]. Inflammation, 2016, 39(6): 2029-2039.
9 Ma J, Wang Y, Wu Q, et al. Seawater immersion aggravates burn-associated lung injury and inflammatory and oxidative-stress responses[J]. Burns, 2017, 43(5): 1011-1020.
10 Li C, Bo L, Li P, et al. Losartan, a selective antagonist of AT1 receptor,attenuates seawater inhalation induced lung injury via modulating JAK2/STATs and apoptosis in rat[J]. Pulm Pharmacol Ther, 2017, 45: 69-79.
11 Ma L, Li Y, Zhao Y, et al. 3,5,4′-tri-O-acetylresveratrol ameliorates seawater exposure-induced lung injury by upregulating connexin 43 expression in lung[J]. Mediators Inflamm, 2013, 2013: 182132.
12 Moraes TJ, Zurawska JH, Downey GP. Neutrophil granule contents in the pathogenesis of lung injury[J]. Curr Opin Hematol, 2006, 13(1): 21-27.
13 Bian Z, Guo Y, Ha B, et al. Regulation of the inflammatory response:enhancing neutrophil infiltration under chronic inflammatory conditions[J]. J Immunol, 2012, 188(2): 844-853.
14 Hudson LD, Hough CI. Therapy for late-phase acute respiratory distress syndrome[J]. Clin Chest Med, 2006, 27(4): 671-677.
15 Liu Z, Zhang B, Wang XB, et al. Hypertonicity contributes to seawater aspiration-induced lung injury: Role of hypoxia-inducible factor 1alpha[J]. EXP LUNG RES, 2015, 41(6): 301-315.
16 Zhang M, Yan X, Liu W, et al. Endothelial semaphorin 7A promotes seawater aspiration-induced acute lung injury through plexin C1 and beta1 integrin[J]. Mol Med Rep, 2017, 16(4): 4215-4221.
17 Guo K, Jin F. NFAT5 promotes proliferation and migration of lung adenocarcinoma cells in part through regulating AQP5 expression[J]. Biochem Biophys Res Commun, 2015, 465(3): 644-649.
18 Han F, Luo Y, Li Y, et al. Seawater induces apoptosis in alveolar epithelial cells via the Fas/FasL-mediated pathway[J]. Respir Physiol Neurobiol, 2012, 182(2-3): 71-80.
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
中學生數理化·七年級數學人教版(2019年10期)2019-11-25 07:33:58
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
文苑(2018年21期)2018-11-09 01:23:06
中學生數理化·高一版(2018年9期)2018-10-09 06:46:50
湖南教育·C版(2018年3期)2018-06-05 16:54:36
中國衛生(2015年9期)2015-11-10 03:11:12
中國衛生(2014年3期)2014-11-12 13:18:12
