分散固相萃取-氣相色譜法測定土壤中的高效氯氰菊酯殘留
2018-03-07 10:25:08蔡曉鈺蔣寶南劉騰飛
上海農業學報 2018年1期
蔡曉鈺,姜 宇,蔣寶南,劉騰飛
(1江蘇省水文水資源勘測局蘇州分局,蘇州215011;2蘇州農業職業技術學院,蘇州215008;3江蘇太湖地區農業科學研究所,蘇州215155
高效氯氰菊酯(β-cypermethrin,β-Cyp),又名戊酸氰醚酯,是典型的Ⅱ型擬除蟲菊酯類殺蟲劑。具有觸殺與胃毒作用,通過改變昆蟲神經膜的通透性,使中毒昆蟲過度興奮、麻痹而死亡[1-2],是國內外廣泛應用的殺蟲劑之一。β-Cyp對蜜蜂、鳥類、魚類、蠶高毒[3-5],對光、熱穩定,在農作物生長過程中長期使用,會造成殘留和累積,帶來嚴重的環境污染與農產品安全問題。土壤是種植農作物的物質基礎,也是農藥在環境中遷移轉化的重要載體、歸宿地和積蓄庫。因此,建立簡單、快速、準確地測定土壤中β-Cyp的檢測方法,對保護土壤環境和農產品安全具有重要意義。
土壤樣品基體復雜,含有色素、甾醇、有機酸等多種干擾物,因此,分析測定前需要經過有效的提取方法和凈化步驟消除基體干擾。目前,土壤中β-Cyp的分析測定,樣品的前處理主要采用傳統的振蕩法[6-8]、勻漿法[9]等提取技術,以及液液分配[7]、固相萃取[7,9]、柱層析[7-8,10-11]等凈化方法,普遍存在操作繁瑣、耗費時間、材料成本高、溶劑使用多等不足。分散固相萃取(Dispersive solid phase extraction,DSPE)[12]是近年來發展起來的一種簡便、快速、高效、經濟的新型樣品前處理技術。它利用直接在試樣提取液中加入的固相吸附劑對基質中干擾雜質的吸附作用,達到除雜凈化的目的。目前在蔬菜、水果等植物性農產品[13-14],肉類、魚類等動物性食品[15-16],以及土壤等環境樣品[17-18]中多種農藥殘留的檢測中已取得了較好的應用效果。但尚未有文獻報道將該技術應用于土壤中β-Cyp的凈化分析。本試驗將DSPE方法應用于土壤樣品前處理,結合氣相色譜-電子捕獲檢測法(GC/ECD),建立一種快速、高效、準確地測定土壤中β-Cyp殘留量的分析方法。
1 材料與方法
1.1 儀器與設備
7890A氣相色譜儀,配電子捕獲檢測器(ECD)(美國Agilent公司);KQ-500DE超聲波清洗器(昆山超聲儀器公司);TG16-WS高速離心機(湖南湘儀實驗儀器公司);HSC-24 B氮吹儀(天津恒奧科技公司);SX2-4-10馬弗爐(上海躍進醫療器械公司);Direct-Q 5 UV超純水機(美國Millipore公司)。
1.2 藥品與試劑
β-Cyp標準溶液,質量濃度100 mg/L,購于農業部環境保護科研監測所;正己烷,色譜純(瑞典Oceanpak公司);N-丙基乙二胺(PSA),40—60μm(中國Agela Technologies公司);十八烷基硅烷鍵合硅膠(C18),40—60μm(美國 Sepax Technologies公司);乙腈、氯化鈉、無水硫酸鎂(620℃灼燒4 h)均為分析純(上海國藥集團化學試劑有限公司)。
1.3 供試土壤
供試土壤取自當地某蔬菜生產基地,采用5點法取樣,取樣深度為0—15 cm。樣品自然風干,除去砂石、草根和其他碎屑,搗碎,充分混勻,四分法取一定量裝入自封袋,備用。制備好的土樣盡快分析,否則于-18℃冷凍保存。
1.4 試驗方法
1.4.1 標準溶液的配制
將100 mg/L的β-Cyp標液從冰箱中取出恢復至室溫,取1 mL置于10 mL容量瓶,用正己烷定容,配成10 mg/L的標準儲備液。使用時再用正己烷稀釋,配制成質量濃度分別為1.0 mg/L、0.5 mg/L、0.2 mg/L、0.05 mg/L和0.02 mg/L的系列標準工作液,貯存于4℃冰箱中,備用。
1.4.2 樣品前處理
1.4.2.1 提取
稱取5.0 g土樣于50 mL聚四氟乙烯離心管中,加入2mL超純水,渦旋混勻,浸潤15min,加入10mL乙腈和2 g氯化鈉混勻,超聲提取15min,加入2 g無水硫酸鎂,渦旋2min,以6 000 r/min離心4min,取上清液待凈化。
1.4.2.2 凈化
稱取150 mg PSA、150 mg C18和300 mg無水硫酸鎂于10 mL離心管中,加入4 mL上述待凈化的提取液,渦旋2 min,以9 000 r/min離心5 min。轉移2 mL上清液,60℃水浴下氮吹近干,定量加入1 mL正己烷溶解殘渣,混勻,過0.22μm有機系濾膜,待GC/ECD測定。
1.4.3 色譜條件
色譜柱:HP-5毛細管柱(30 m×0.32 mm×0.25μm);檢測器溫度300℃;進樣口溫度250℃;柱箱溫度:初始溫度180℃,保持2 min,以12℃/min的速率升溫至260℃,保持7 min;載氣:高純氮氣,流速2.8 mL/min;尾吹氣:60 mL/min;進樣量 1μL,不分流進樣。
1.4.4 添加回收試驗
稱取5.0 g土壤空白樣品3份,分別添加10 mg/L的標準儲備液500μL、100μL及1 mg/L的標準工作溶液100μL,添加水平分別相當于1.0 mg/kg、0.2 mg/kg、0.02 mg/kg,充分混勻,靜置1 h讓溶劑揮發,按前述方法提取、凈化和GC/ECD測定,對每個添加水平作3個平行,考察方法的準確度和精密度。
1.4.5 基質效應分析
將土壤空白樣品按本試驗方法進行前處理,用該空白土壤提取凈化液作溶劑,配制系列基質標準溶液。取基質標準溶液與溶劑標準溶液,用GC/ECD進樣測定,以標準溶液的質量濃度(x)為橫坐標,峰面積響應值(y)為縱坐標,分別繪制基質標準曲線和溶劑標準曲線。
2 結果與討論
2.1 提取溶劑的選擇
文獻報道一般采用乙腈[6,9]、丙酮-石油醚[7,11]等對土壤中的 β-Cyp進行提取。本試驗在空白土壤中添加 100μL供試農藥的標準儲備液,比較了乙腈、丙酮-石油醚(1∶1,V/V)、丙酮-石油醚(3∶1,V/V)對樣品中β-Cyp的提取效果。結果表明(表1):以乙腈作為提取溶劑時β-Cyp的回收率最高,而且乙腈對土壤基質中的蠟類、脂肪等非極性成分提取能力弱,提取液雜質較少,顏色較淺,有利于簡化后續的凈化操作,故選擇乙腈作為提取溶劑。

表1 不同提取溶劑的回收率結果Table 1 Recoveries of different extraction solvents(n=3) %
2.2 提取方式的選擇
超聲波法利用超聲波的空化作用和熱作用,可以加速分析物從樣品基體中溶解出來,具有操作簡單、節省時間、提取效率高的特點,可以很好地用于批量樣品的同時處理中,故選用超聲波法提取樣品。在空白土壤中添加100μL供試農藥的標準儲備液,比較了5 min、10 min、15min、20min、30min的超聲時間對提取效果的影響。結果表明(圖1):超聲時間為5min、10 min時,β-Cyp提取回收率偏低,低于80%,延長超聲時間至15 min時,回收率有所增高,達到91.5%,進一步延長提取時間,回收率變化不大。為節省樣品提取時間,選擇超聲時間為15min。
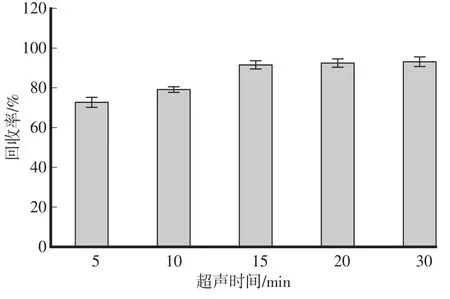
圖1 不同超聲時間對土壤中β-Cyp提取回收率的影響(n=3)Fig.1 Effect of ultrasonic time durations on extraction efficiencies ofβ-Cyp in soil(n=3)
2.3 凈化條件的選擇
采用DSPE的方式凈化樣品,并選用PSA和C18作為分散固相吸附劑,PSA用于去除土壤提取液中的脂肪酸、金屬離子、極性色素等干擾物質,C18用于去除甾醇、油脂和弱極性的色素。取4.0 mL空白土壤提取液,加入100μL供試農藥的標準儲備液,混勻后經PSA和C18吸附劑渦旋凈化,比較了不同用量吸附劑對提取液的凈化效果及對目標農藥回收率的影響。結果表明(表2):隨著PSA和C18用量的增加,提取液的凈化效果提高,顏色逐漸變淺,但同時目標物β-Cyp的回收率緩慢降低。當PSA和C18用量均為150 mg時,β-Cyp的回收率較好,且此時提取液的顏色較淺,色譜分析時的干擾峰較少。故選擇分散吸附劑PSA和C18的用量各為150 mg,同時在提取液中加入300 mg的無水硫酸鎂以除去溶劑中殘留的少量水分,避免PSA粉末吸水后性能下降。

表2 β-Cyp經C18和PSA吸附劑處理后的回收率結果Table 2 Recoveries ofβ-Cyp after purified by C18 and PSA
2.4 色譜分析結果
在本方法色譜條件下,對β-Cyp標準溶液、土壤空白樣品及空白加標樣品進行測定,對應的色譜圖見圖2—圖4。從色譜圖中可以看出,β-Cyp的保留時間約為10.89 min,峰形良好,無拖尾現象,土樣經前處理后,基質中的內源性雜質對其測定沒有干擾,說明本方法的色譜條件及前處理條件選擇合適。
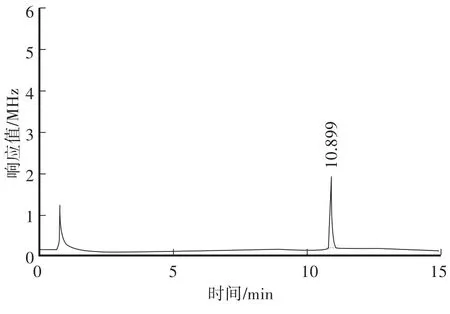
圖2 β-Cyp標準溶液(0.2 mg/L)Fig.2 GC ofβ-Cyp standard solution(0.2 mg/L)
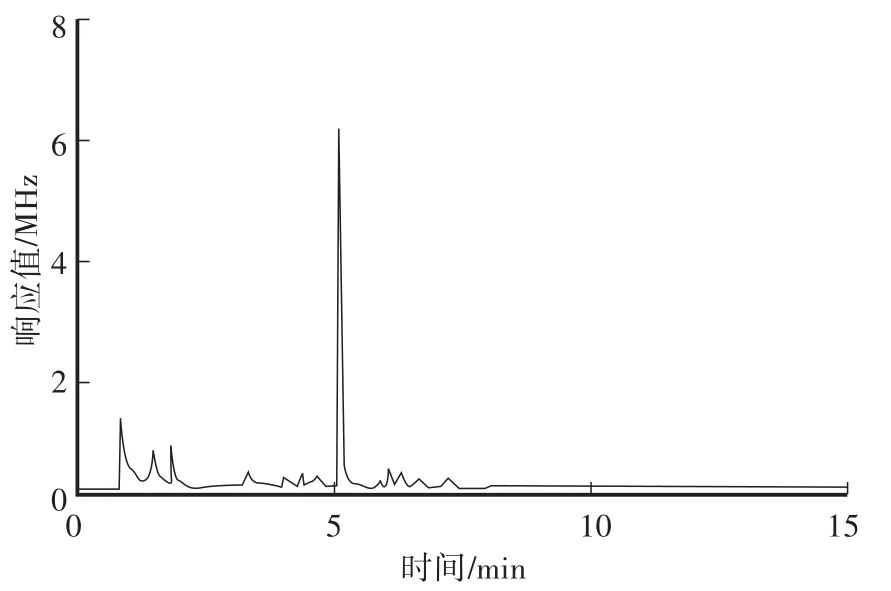
圖3 土壤空白樣品色譜圖Fig.3 GC of a blank soil samp le
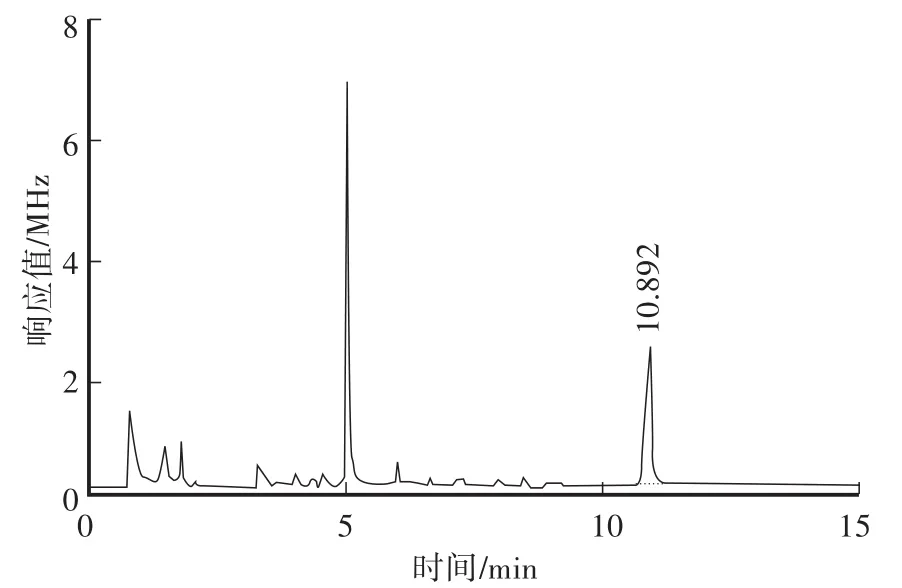
圖4 土壤空白加標色譜圖(0.2 mg/kg)Fig.4 GC of 0.2 mg/kg spiked soil sam p le
2.5 方法線性范圍與標準曲線
取配制好的質量濃度為0.02—1.0 mg/L的β-Cyp系列溶劑標準溶液和基質標準溶液進行GC/ECD測定,以標準溶液的質量濃度(x)為橫坐標,對應峰面積(y)為縱坐標,繪制標準曲線。
表3為β-Cyp在溶劑和基質中的標準曲線和相關系數。由表3可以看出,β-Cyp的質量濃度(x)在0.02—1.0 mg/L范圍內與峰面積(y)呈良好的線性關系,相關系數均大于0.9930。

表3 β-Cyp的標準曲線與相關系數Table 3 Standard curve equations and correlation coefficients
2.6 基質效應分析
在氣相色譜分析中,基質效應可引起分析結果的偏差和對樣品分析過程回收率的錯誤計算[19],所以對基質效應考察評估并采取有效措施進行消除或補償,是進行農藥殘留準確分析的重要環節。本試驗采用基質標準曲線和溶劑標準曲線的斜率之比(k)評價基質效應[20]:當k在0.9—1.1時,基質效應不明顯;當k大于1.1時,為基質增強效應;當k小于0.9時,為基質減弱效應。
根據表3結果,得出β-Cyp的k值為1.3867,表明采用本方法檢測土壤葉中的β-Cyp時存在基質增強效應。因此,在使用外標法定量時,采用空白土壤樣品的凈化液稀釋標樣,以消除基質干擾,減少誤差。
2.7 方法準確度與精密度
表4為土壤中β-Cyp的添加回收率和相對標準偏差。從表4可以看出,當添加濃度為0.02—1.0 mg/kg時,β-Cyp在土壤中的平均回收率為83.8%—100.6%,相對標準偏差(n=3)為3.7%—9.8%,表明該方法具有很好的準確性和精密性,符合農藥殘留檢測的要求[21]。
2.8 方法檢出限與定量限
在本方法條件下,對β-Cyp最小添加水平(0.02mg/kg)的土壤樣品進行測定,以色譜圖中信噪信號的3倍為檢出限,以最小添加水平為定量限,得到土壤中β-Cyp的檢出限和定量限分別為0.003mg/kg、0.02mg/kg。
2.9 實際樣品分析
利用本試驗建立的分析方法對當地蔬菜生產基地采集的土壤樣品進行檢測。結果顯示,在被檢測的7份土壤樣品中,共檢出3份土壤樣品含有β-Cyp,含量分別為0.0253 mg/kg、0.0304mg/kg、0.0343mg/kg。通過調查,該蔬菜生產基地在取樣前曾施用了4.5%高效氯氰菊酯乳油防治青菜和莧菜上的菜青蟲,造成了β-Cyp在土壤中的殘留,與農藥殘留分析結果相吻合。

表4 土壤中β-Cyp的添加回收率和相對標準偏差Table 4 Recoveries and relative standard deviations ofβ-Cyp in soil samp le(n=3)
3 結論
本試驗以乙腈為提取溶劑,采用超聲波法提取,PSA和C18混合吸附劑分散萃取凈化,GC/ECD檢測,建立了土壤中β-Cyp殘留量的分析方法。方法的準確度、精密度和靈敏度均滿足殘留分析的要求。在0.02—1.0 mg/kg添加水平下,β-Cyp在土壤中的平均回收率為83.8%—100.6%,相對標準偏差小于10%,檢出限、定量限分別為0.003 mg/kg、0.02 mg/kg。
該方法操作簡單,定量準確,溶劑用量少,對檢測條件要求低,易于普及掌握,為土壤中β-Cyp殘留的研究提供了可靠的分析手段,對保證農產品安全具有重要意義。
[1]馬萍,秦龍娟,張亞然,等.高效氯氰菊酯對小鼠肝細胞的氧化損傷[J].環境科學學報,2012,32(3):757-761.
[2]SODERLUND DM,NIPPLE DC.Themolecular biology of knockdown resistance to pyrethroid insecticides[J].Insect Biochem Mol Biol,2003(33):563-577.
[3]王健,劉麗麗,余凱敏,等.高效氯氰菊酯對斑馬魚胚胎毒性的研究[J].生物技術通報,2014,(10):223-229.
[4]李前龍,唐旭東,徐莉,等.不同溫度下5種擬除蟲菊酯類農藥對家蠶的毒性變化[J].蠶業科學,2013(1):70-75.
[5]許迪,潘竟林,劉萬強,等.多殺菌素、阿維菌素乳油和高效氯氰菊酯3種農藥對環境生物的安全性評價[J].生態毒理學報,2013,8(6):897-902.
[6]何華,徐存華,孫成,等.高效氯氰菊酯在土壤中的降解動態[J].中國環境科學,2003,23(5):490-492.
[7]代雪芳,毛佳,宋爽,等.高效氯氰菊酯在苜蓿和土壤中的殘留及消解動態[J].西南農業學報,2014,27(2):658-663.
[8]盧洪秀,陳俊,馬步春.混合農藥在甘藍及土壤中的殘留動態研究[J].上海農業學報,2014,30(3):82-88.
[9]王彥輝,柏連陽,李欣,等.高效氯氰菊酯在柑橘和土壤中的殘留消解動態[J].農藥,2011,50(6):428-430.
[10]丁蕊艷,陳子雷,楊國生.凝膠滲透色譜凈化-氣相色譜法測定高效氯氰菊酯在棉花及土壤中的殘留[J].農藥,2012,51(11):822-824.
[11]王飛,王軍,李純.高效氯氰菊酯在蘋果和土壤中的消解動態及殘留安全性評價[J].安徽農業科學,2010,38(20):10837-10838.
[12]ANASTASSIADESM,LEHOTAY S J,STAJNBAHER D,et al.Fast and easymultiresiduemethod employing acetonitrile extraction/partitioning and“dispersive solid-phase extraction”for the determination of pesticide residues in produce[J].JAOAC Int,2003,86(2):412-431.
[13]KAEWSUY A P,BREWERW E,WONG J,et al.Automated QuEChERS tips for analysis of pesticide residues in fruits and vegetables by GCMS[J].JAgric Food Chem,2013,61(10):2299-2314.
[14]ALBERT A,KRAMER A,SCHEEREN S,et al.Rapid and quantitative analysis of pesticides in fruits by QuEChERS pretreatment and lowtemperature plasma desorption/ionization orbitrap mass spectrometry[J].Anal Methods,2014,15(15):5463-5471.
[15]蘇明明,董振霖,徐靜,等.QuEChERS法聯合在線凝膠過濾色譜-氣相色譜-質譜聯用法快速測定魚肉中16種農藥殘留量[J].食品安全質量檢測學報,2014,5(6):1757-1764.
[16]郭禮強,宮小明,丁葵英,等.基于QuEChERS提取的液相色譜-串聯質譜法測定干腌火腿中15種真菌毒素[J].分析測試學報,2015,34(2):141-146.
[17]劉騰飛,楊代鳳,董明輝,等.茶園土壤中有機磷農藥的分散固相萃取-氣相色譜測定[J].上海農業學報,2016,32(2):70-74.
[18]AKAMATSU M,TSUJITA K,PITIYONT V,et al.Pesticide residue analyses of soils collected from suburban agricultural fields around Bangkok[J].Trop Agr Develop,2013,57(1):8-15.
[19]劉騰飛,張麗,楊代鳳,等.茶園土壤中擬除蟲菊酯類農藥殘留檢測[J].江蘇農業學報,2015,31(4):935-941.
[20]蔣寶南,劉騰飛,單建明,等.QuEChERS-GC/μECD法測定土壤中的毒死蜱殘留量[J].江蘇農業科學,2014,42(12):332-335.
[21]中華人良共和國農業部.中華人民共和國農業行業標準-農藥殘留試驗準則NY/T788-2004[S/J].農產品質量與安全,2004(4):29-33.
