木荷優樹無性系種質SSR標記的遺傳多樣性分析*
2017-06-23 12:08:48楊漢波王幫順徐肇友陳煥偉周志春
林業科學 2017年5期
關鍵詞:分析
楊漢波 張 蕊 王幫順 徐肇友 陳煥偉 周志春
(1. 中國林業科學研究院亞熱帶林業研究所 浙江省林木育種技術研究重點實驗室 杭州 311400; 2. 浙江省龍泉市林業科學研究院 龍泉 323700)
?
木荷優樹無性系種質SSR標記的遺傳多樣性分析*
楊漢波1張 蕊1王幫順2徐肇友2陳煥偉2周志春1
(1. 中國林業科學研究院亞熱帶林業研究所 浙江省林木育種技術研究重點實驗室 杭州 311400; 2. 浙江省龍泉市林業科學研究院 龍泉 323700)
【目的】 利用SSR標記深入研究木荷優樹無性系種質的遺傳多樣性,揭示其遺傳多樣性地理分布特點及種質間遺傳關系,為木荷種質資源的保護和育種親本的選擇提供理論依據。【方法】 利用10對SSR引物,分析我國5個省份24個地區的734份木荷優樹無性系種質的遺傳多樣性和遺傳結構。利用CERVUS、GenAIEx 6.5、NTSYS、Arlequin和STRUCTURE 2.3軟件進行無效等位基因檢測、遺傳參數估算、主坐標分析、聚類圖構建、遺傳變異分析及遺傳結構分析。【結果】 10對引物共檢測到105個等位基因(Na),平均每個引物為10.5個,ss16引物檢測到的等位基因數最多,為16個。Shannon’s信息指數(I)變化范圍為1.121~1.908,平均值為1.473; 多態信息指數(PIC)范圍為0.557~0.807,平均值為0.668; 平均期望雜合度(He)和觀測雜合度(Ho)分別為0.713和0.735。木荷優樹無性系種質的主坐標(PCoA)和遺傳結構分析基本可以保持一致,供試734份木荷優樹無性系種質可被分為3個PCoA類群,而在遺傳結構上可劃分為5個群組。24個種質群體間遺傳距離范圍為0.030~0.804,平均為0.230,表明群體間的親緣關系較近,但仍有部分種質群體間存在較遠的親緣關系,如HNSZ和GDSX,JXFY和FJSX等; 不同種質群體Shannon’s信息指數(I)變化范圍為0.980~1.431,遺傳多樣性與地理分布不完全相關。STRUCTURE分析表明,71.1%的木荷優樹無性系種質遺傳組分相對比較單一,28.9%的種質遺傳背景比較復雜。分子方差分析(AMOVA)表明,供試的木荷優樹無性系種質有5.91%的遺傳變異存在于群體間,而94.09%的遺傳變異來自于群體內。【結論】 木荷優樹無性系種質存在豐富的遺傳多樣性,各群體間遺傳多樣性水平相差較大。在木荷雜交育種親本選配時不僅要考慮地理遠緣,還應考慮親本群體(個體)間的親緣關系。
木荷; 優樹; SSR標記; 遺傳多樣性; 遺傳結構
種質資源的收集是林木育種的關鍵環節。截止2013年底,我國已收集保存各類林木種質資源16萬份。多樣性的評價和遺傳背景的研究是種質資源利用的前提(Chuanfuetal., 2008)。許多學者借助表型性狀變異開展植物種質資源遺傳多樣性研究(王永康等, 2014; Zekaetal., 2015)。然而,由于某些性狀受環境因素和生長期的影響,基于形態和生長性狀對種質資源遺傳多樣性的研究具有一定的局限性,不能準確反映種質資源個體間的遺傳差異和親緣關系(Terzopoulosetal., 2008)。自20世紀90年代以來,具有共顯性、高分辨率和重復性好等特點的SSR(Simple Sequence Repeat)標記技術已廣泛應用于作物及多年生木本植物的遺傳多樣性和遺傳結構分析中,并取得了一系列的成果(Ferr?oetal., 2015; 趙爽等, 2016; Lassoisetal., 2016)。另外,在種質親緣關系研究中,主坐標分析(PCoA)與STRUCTURE分析方法的結合已得到許多應用。對于遺傳標記結果,主坐標分析(PCoA)通過相似性進行個體區分,可以清晰地顯示個體間的相互關系; STRUCTURE分析采用混合模型,可以對所有個體進行劃分,反映群體的遺傳結構本質,還可以根據個體等位基因的組分數,推斷群體中具有復雜遺傳背景的個體或者發生遷移的個體,對群體中的個體進行歸屬判斷(Sunetal., 2003; 宗緒曉等, 2010)。如在林木親緣關系研究中,采用主坐標(PCoA)與STRUCTURE 2種分析方法相結合處理鵝掌楸(Liriodendronchinense)(楊愛紅等, 2014)、紅椿(Toonaciliata)(李培等, 2016)等分子標記數據,得到了更為準確、豐富的遺傳多樣性與遺傳結構分析結果。
木荷(Schimasuperba)為山茶科(Theaceae)木荷屬(Schima)常綠大喬木,自然分布于31°N以南105°E以東的廣大地區,是該區域常綠闊葉林的主要建群種。木荷早期速生,材性優良,抗逆性強,亦是我國南方各省區的珍貴優質闊葉用材和高效生物防火樹種,在商品用材林和生態防火林建設中占有重要地位(張蕊等, 2013; 楚秀麗等, 2014),以珍貴優質用材和高效生物防火為目標的木荷育種研究已成為林木育種學家研究的重點。作者所在研究組自2001年開展木荷育種工作以來,經過不斷搜集、補充,目前已保存國內5個省份24個地區的木荷優樹無性系種質734份,并借助表型標記開展了木荷地理種源遺傳變異研究(張萍等, 2004; 周志春等, 2006; 王秀花等, 2011)。本研究在辛娜娜等(2015)對部分木荷育種親本進行遺傳多樣性分析的基礎上,利用SSR標記進一步對搜集保存的全部木荷優樹無性系種質進行遺傳多樣性分析,擬通過大規模樣本間的遺傳多樣性及遺傳結構的詳細系統比較和分析,全面揭示我國木荷優樹無性系種質遺傳多樣性地理分布特點和種質群體(個體)間的遺傳關系,為我國木荷優樹無性系種質的深入研究和開發利用提供科學依據,同時也為我國木荷的長期多目標育種奠定基礎。
1 材料與方法
1.1 試驗材料
研究材料來源于浙江省龍泉市林業科學研究院上圩基地木荷優樹無性系種質基因庫(28°03′N, 119°06′E)。基地面積為6.7 hm2,海拔200~300 m,相對濕度79%,年均降雨量1 664.8~1 706.2 mm。2010年至今,在浙江、江西、福建等地選擇優樹1 000余株,嫁接保存734份。選優林分要求林齡20年、面積1.0 hm2以上,以木荷為主的優良天然林或起源明確的人工林; 優樹選擇條件為樹型高大,干形通直圓滿,生長量明顯高于附近3~5株同齡優勢木等。以本地1~2年生木荷容器苗為砧木,在3—4月份選用帶有休眠芽的穗條,采用切接的方法嫁接優樹無性系,在苗圃地集中培育2年生大容器嫁接苗,然后根據木荷優樹無性系種質基因庫配置圖(株行距為4 m×4 m)將嫁接苗移栽定植至基因庫相應的位置上。將所有木荷優樹無性系種質按照產地來源分為24個種質群體(表1), 2015年6月,對全部734份木荷優樹無性系種質采集其頂端新發枝條上的新鮮嫩葉,將其放入液氮中帶回實驗室,置于-80 ℃冰箱中保存備用。
表1 木荷優樹無性系種質來源
Tab. 1 Origin ofSchimasuperbagermplasms

群體Population數量Number來源OriginGDXS32廣東各縣市GuangdongHNSZ22湖南桑植Sangzhi,HunanHNCB30湖南城步Chengbu,HunanHNQY23湖南祁陽Qiyang,HunanJXSY48江西上猶Shangyou,JiangxiJXTG5江西銅鼓Tonggu,JiangxiJXCY21江西崇義Chongyi,JiangxiJXLN21江西龍南Longnan,JiangxiJXYT10江西鷹潭Yingtan,JiangxiJXXF56江西信豐Xinfeng,JiangxiJXFY7江西分宜Fenyi,JiangxiZJSC24浙江遂昌Suichang,ZhejiangZJLQ57浙江龍泉Longquan,ZhejiangZJQY28浙江慶元Qingyuan,ZhejiangFJLC48福建連城Liancheng,FujianFJNP57福建南平Nanping,FujianFJYX54福建尤溪Youxi,FujianFJFZ9福建福州Fuzhou,FujianFJYA30福建永安Yong’an,FujianFJSW21福建邵武Shaowu,FujianFJJO65福建建甌Jian’ou,FujianFJGT49福建古田Gutian,FujianFJSX9福建沙縣Shaxian,FujianFJSC8福建順昌Shunchang,Fujian總計Total734
1.2 基因組DNA提取及SSR擴增
采用試劑盒提取木荷基因組DNA,1.0%瓊脂糖凝膠電泳檢測DNA純度和完整性,NanoDrop-2000超微量分光光度計(Thermo,美國)檢測其濃度,最后稀釋成20 ng·μL-1,-20 ℃保存備用。本研究選用13對條帶清晰、多態性強的SSR引物進行PCR擴增(辛娜娜等, 2015)。PCR擴增反應在TaKaRa PCR Thermal Cycler Dice Touch上進行。PCR擴增反應體系25 μL: 含12.5 μL 2×Taq Plus Master Mix, 10 μmol·L-1上下游引物和50 ng基因組DNA。PCR擴增程序為: 95 ℃預變性5 min; 94 ℃變性30 s,53 ℃退火30 s,72 ℃延伸45 s(35個循環); 72 ℃延伸10 min (辛娜娜等, 2015)。擴增產物在Qsep100TM全自動核酸蛋白自動分析系統上進行電泳分離檢測和片段大小的測定,Qsep100TM能以1~4 bp的分辨率高效區分20~20 000 bp DNA片段 (http://www.sciencemag.org/content/348/6241/1383.1.full)。
1.3 遺傳參數分析
對SSR檢測結果進行峰圖分析及等位基因的讀取,然后采用Cervus 3.0.7軟件(Kalinowskietal., 2007)計算多態信息含量(PIC),并檢測各SSR位點的無效等位基因(null alleles)頻率,去除無效等位基因頻率超過0.2的引物組合,以確保后續遺傳多樣性分析結果的準確性(文亞峰等, 2013)。利用GenAlEx6.502軟件(Peakalletal., 2012)對結果進行遺傳多樣性分析。分別計算: 1)等位基因數(Na); 2)有效等位基因數(Ne); 3)Shannon’s信息多樣性指數(I); 4)觀測雜合度(Ho)與期望雜合度(He); 5)群體間Nei’s遺傳相似系數和遺傳距離,并利用NTSYS pc 2.1軟件(Rohlf , 2000)繪制基于UPGMA法的樹狀聚類圖。利用GenAlEx6.502軟件,計算各木荷優樹無性系種質個體間的遺傳距離,并進行主坐標(PCoA)分析。用Arlequin軟件對群體間和種群內分子遺傳變異進行AMOVA分子方差分析(Excoffieretal., 2010)。
1.4 群體遺傳結構和模擬聚類分析
利用Genepop 4.5軟件(Rousset, 2008),采用馬爾科夫鏈方法(Markov chain method,MC)對SSR位點之間的關聯性進行顯著性檢驗,得到無偏估計P值,當P<0.05表明連鎖不平衡具有顯著性。采用軟件STRUCTURE 2.3(Pritchardetal., 2000)進行木荷優樹無性系種質遺傳結構分析。這一軟件分析目的在于找到個體合適的分組數,K值。首先確定K值取值范圍為1~20,然后運行軟件,對每個K值進行個體分組分析,每個K值重復運行100次,參數iterations和burn-in period均設為10 000。再根據Evanno等(2005)提供的ΔK方法確定合適的分組K值。
2 結果與分析
2.1 SSR位點多態性分析
13對SSR引物中,有7對引物的無效等位基因(null alleles)頻率為正值,其中ss01、ss24和ss30的無效等位基因頻率均大于0.2,并且顯著偏離哈迪-溫伯格平衡(表2),因此,在后續遺傳多樣性分析中去除這3個位點以確保結果的準確性。篩選出的10對引物組合能準確獲得不同材料在不同位點的等位基因片段大小及相應的電泳峰圖(圖1)。10對SSR引物在734份木荷優樹無性系種質中擴增出105個等位基因(Na),平均每對引物擴增出10.5個等位基因,有效等位基因數(Ne)為3.756(表2)。 10對木荷SSR引物間存在較大的差異,其中,等位基因數(Na)最多的引物是ss16,為16個,其有效等位基因數(Ne)為4.245; 其次是ss32、ss22,Na均為14個,Ne分別為5.874個和4.868個;Na最少的是引物ss02和ss13,均為6個,Ne分別為2.602個和2.677個。10對SSR引物的多態信息含量(PIC)范圍為0.557~0.807,平均值為0.668,表明所選的SSR引物在木荷優樹無性系種質上多態性豐富。期望雜合度(He)和觀測雜合度(Ho)的變化范圍分別為0.616(ss02)~0.830(ss32)和0.431(ss02)~0.965(ss22),平均分別為0.713和0.735。Shannon’s信息多樣性指數(I)變化范圍為1.121~1.908,平均為1.473,這表明木荷優樹無性系種質具有較豐富的遺傳多樣性。
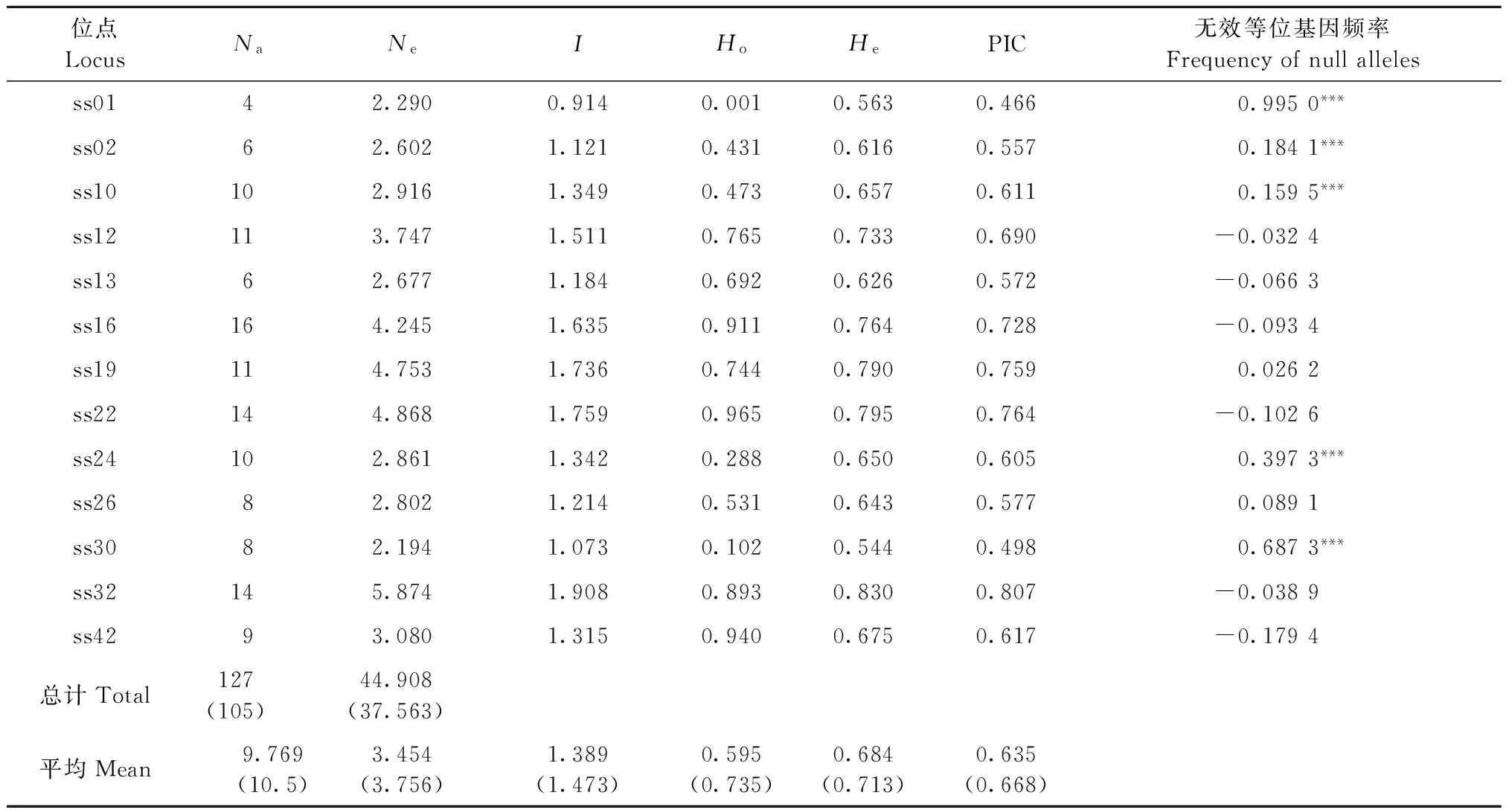
表2 13對SSR引物在全部參試種質中的遺傳多樣性參數①Tab. 2 The genetic diversity parameters of 13 SSR primers in all tested collections of Schima superba
①Na: 等位基因數;Ne: 有效等位基因數;I: Shannon’s信息指數;Ho: 觀測雜合度;He: 期望雜合度; PIC: 多態信息含量。下同。括號內為去除無效等位基因頻率大于0.2的SSR引物后的10對引物的遺傳多樣性參數。*** :P<0.001。
Na: Number of different alleles;Ne: Number of effective alleles;I: Shannon’s information index;Ho: Observed heterozygosity;He: Expected heterozygosity; PIC: Polymorphic information content. The same below. The parameters in brackets are the parameters of genetic diversity for remainder 10 primers which removing the primers of which the frequency of null alleles is higher than 0.2. *** :P<0.001.
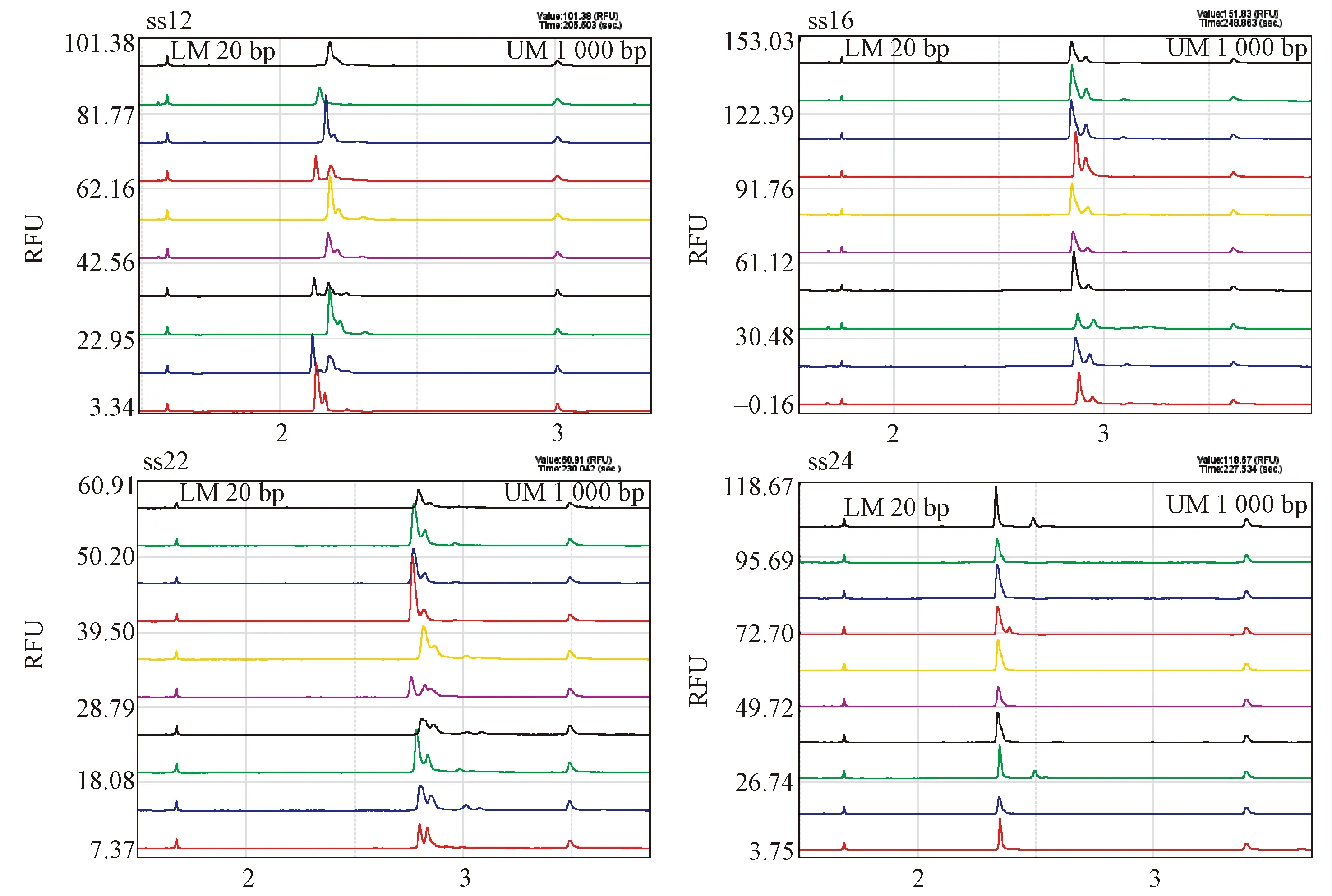
圖1 引物ss12,ss16,ss22和ss24在部分材料中的電泳圖譜Fig.1 The part of electrophoretogram of primer ss12, ss16, ss22 and ss24
2.2 群體遺傳變異及結構分析
24個種質群體的有效等位基因數(Ne)變化范圍較大,在2.593(HNSZ)~3.647(FJJO)之間(表3)。各參試群體的Shannon’s信息指數(I)變化在0.980(JXFY)~1.431(FJJO)之間,平均為1.246。觀測雜合度(Ho)最小的是 HNSZ(0.545),最大的是JXTG(0.820), JXLN次之(0.781),平均為0.725。期望雜合度(He)最小的是HNSZ(0.516),最大的是FJJO(0.709),平均為0.645。部分群體的有效等位基因數(Ne)和Shannon’s信息指數(I)的變化趨勢不一致,如FJNP的Ne高于FJYX,而二者的I值均為1.338,HNCB的Ne高于FJYX,但I卻低于FJYX。同樣,部分群體的觀察雜合度(Ho)和期望雜合度(He)也表現出這種不一致的變化趨勢。AMOVA分析結果(表4)顯示,木荷優樹無性系種質群體間的遺傳變異為5.91%,而群體內遺傳變異顯著,為94.09%。
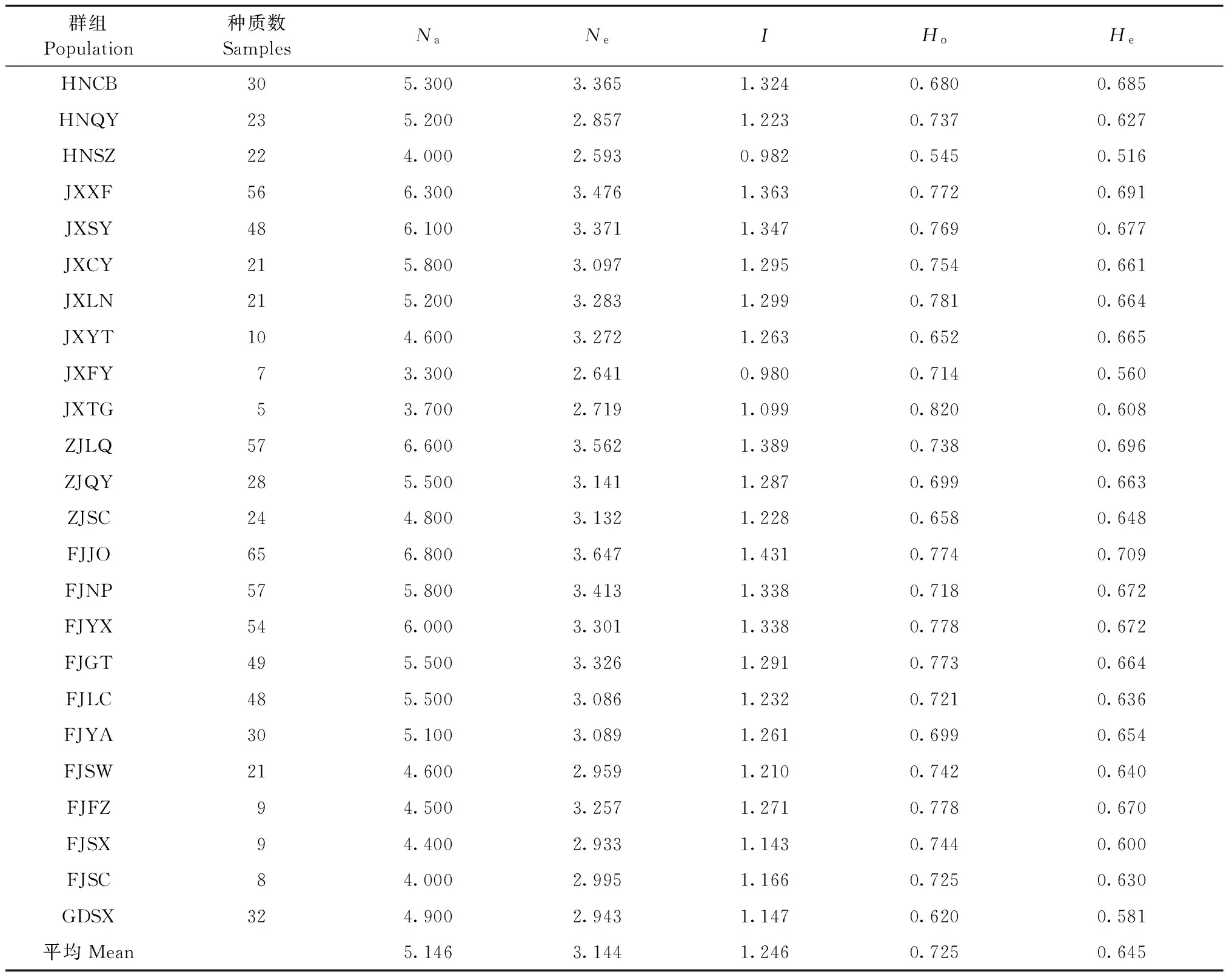
表3 24個木荷優樹無性系種質群體的遺傳多樣性Tab. 3 Genetic diversity of 24 Schima superba plus tree clone populations
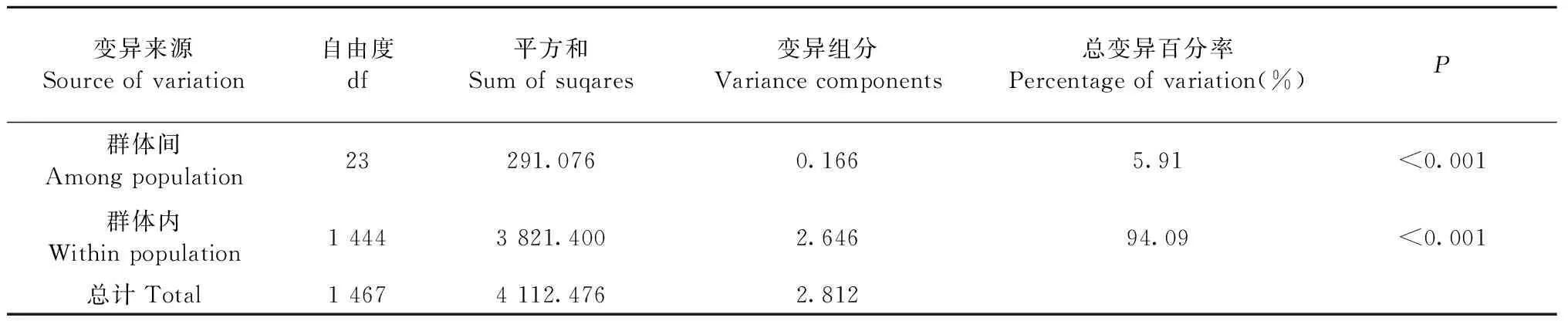
表4 24個種質群體的AMOVA分析Tab. 4 Analysis of molecular variance for 24 germplasm populations
連鎖不平衡(LD)檢測結果顯示各SSR位點間不存在連鎖,處于完全獨立狀態(表5)。STRUCTURE分析中,ΔK在K=5時有明顯的峰(圖2),因此將734份木荷優樹無性系種質分為5個群組,并繪制出遺傳結構圖(圖3)。分析各木荷優樹無性系種質在不同群組的Q值,發現522份種質(71.1%)在某一群組中的Q值大于0.5,推測其遺傳組分相對比較單一,被劃分到5個群組中的1個,而剩余的212份(28.9%)在5個群組中Q值均小于0.5,沒有明確的群組歸屬特性,形成了1個混合群(mixed group)(表6)。GDSX中的96.9%(31份)歸屬于1個群組(綠色群組),Q平均值為0.88,僅1份種質劃分到混合群,表明該群體遺傳組分相對單一。來自FJSC(8份,100%)、FJSX(8份,88.9%)和HNSZ(22份,100%)的大部分種質被歸為1個群組(黃色群組),表明它們的遺傳結構相似。參試木荷優樹無性系種質遺傳結構與地理分布不完全相關,如JXSY、FJNP和JXXF等群體的種質材料在5個群組中均有分布。
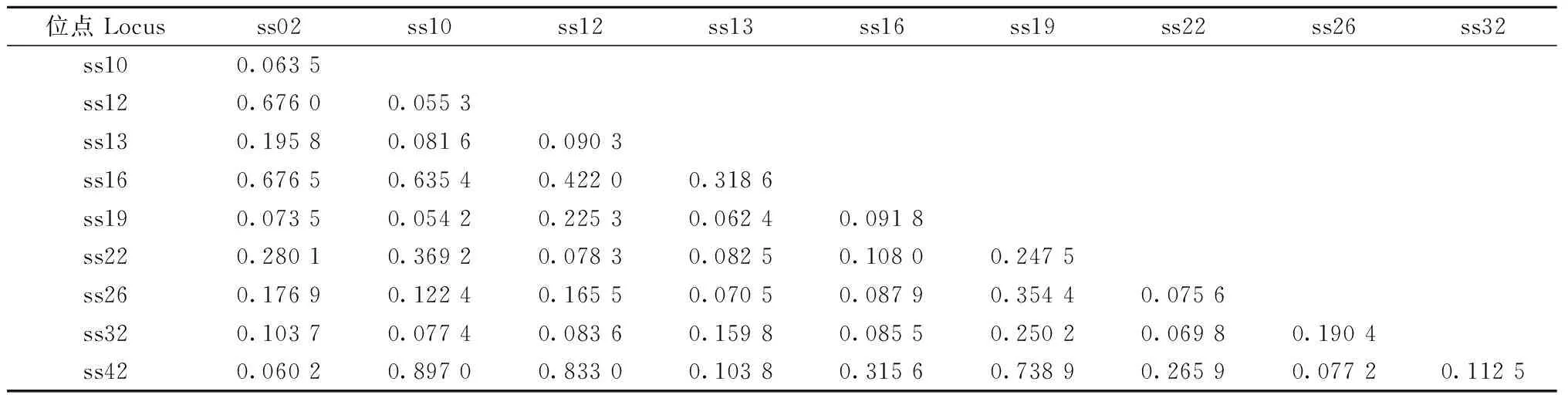
表5 10個SSR位點的連鎖不平衡檢驗結果(P值)Tab. 5 Linkage disequilibrium test result of 10 SSR loci (P value)
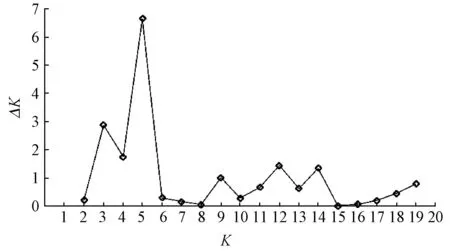
圖2 待定群體數K與估計值ΔK的關系Fig.2 Relations between the number of determined group K and estimated value ΔK
2.3 親緣關系分析
主坐標(PCoA)分析結果顯示,734份木荷優樹無性系種質可分為A、B和C 3個類群(圖4)。A類群所包含的種質數量最多,占全部種質的41%,B、C 2個類群所包含的種質數量差異不大,分別占材料總數的29.7%和29.3%。5個省(市)的種質在3個類群中均有分布,3個類群間有少量相互滲透的現象。A類群的典型代表是HNSZ(20份)、ZJSC(14份)和ZJQY(16份)等, B類群的典型代表是FJSW(13份)、HNCB(16份)和 FJFZ(4份)等,C類群的典型代表有FJYX(37份)、JXFY(4份)和GDSX(27份)等。總體上看,福建、浙江和江西的資源在大范圍內呈分散分布,說明其遺傳背景寬廣。基于兩兩個體間遺傳距離的主坐標(PCoA)分析與遺傳結構分析聚類結果基本可以保持一致: A類群與粉紅色和黃色群組對應,B類群與紅色群組和藍紫色群組相對應,C類群與綠色群組對應; 群體結構分析中劃分到混合群體的材料在PCoA分析中也被劃分到與Q值最大的類群所對應的區域,或與該區域較近的位置。

圖3 參試木荷優樹無性系種質遺傳結構分組Fig.3 Estimated population STRUCTURE for Schima superba resources populations圖中5種顏色表示5個不同的群組,每條彩色豎線代表一份種質,不同顏色所占比例越大,則該種質被劃分到相應群組的可能性就越大。Each germplasm is represented by a single color line; there are five population groups; the more proportion of the color, the more possibility of the represented germplasm by the color divided into the corresponding population.
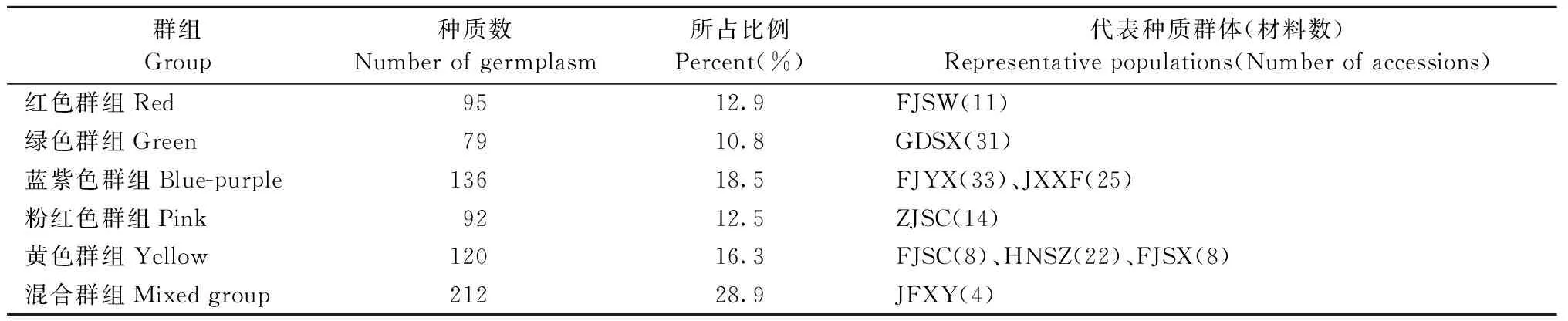
群組Group種質數Numberofgermplasm所占比例Percent(%)代表種質群體(材料數)Representativepopulations(Numberofaccessions)紅色群組Red9512.9FJSW(11)綠色群組Green7910.8GDSX(31)藍紫色群組Blue-purple13618.5FJYX(33)、JXXF(25)粉紅色群組Pink9212.5ZJSC(14)黃色群組Yellow12016.3FJSC(8)、HNSZ(22)、FJSX(8)混合群組Mixedgroup21228.9JFXY(4)
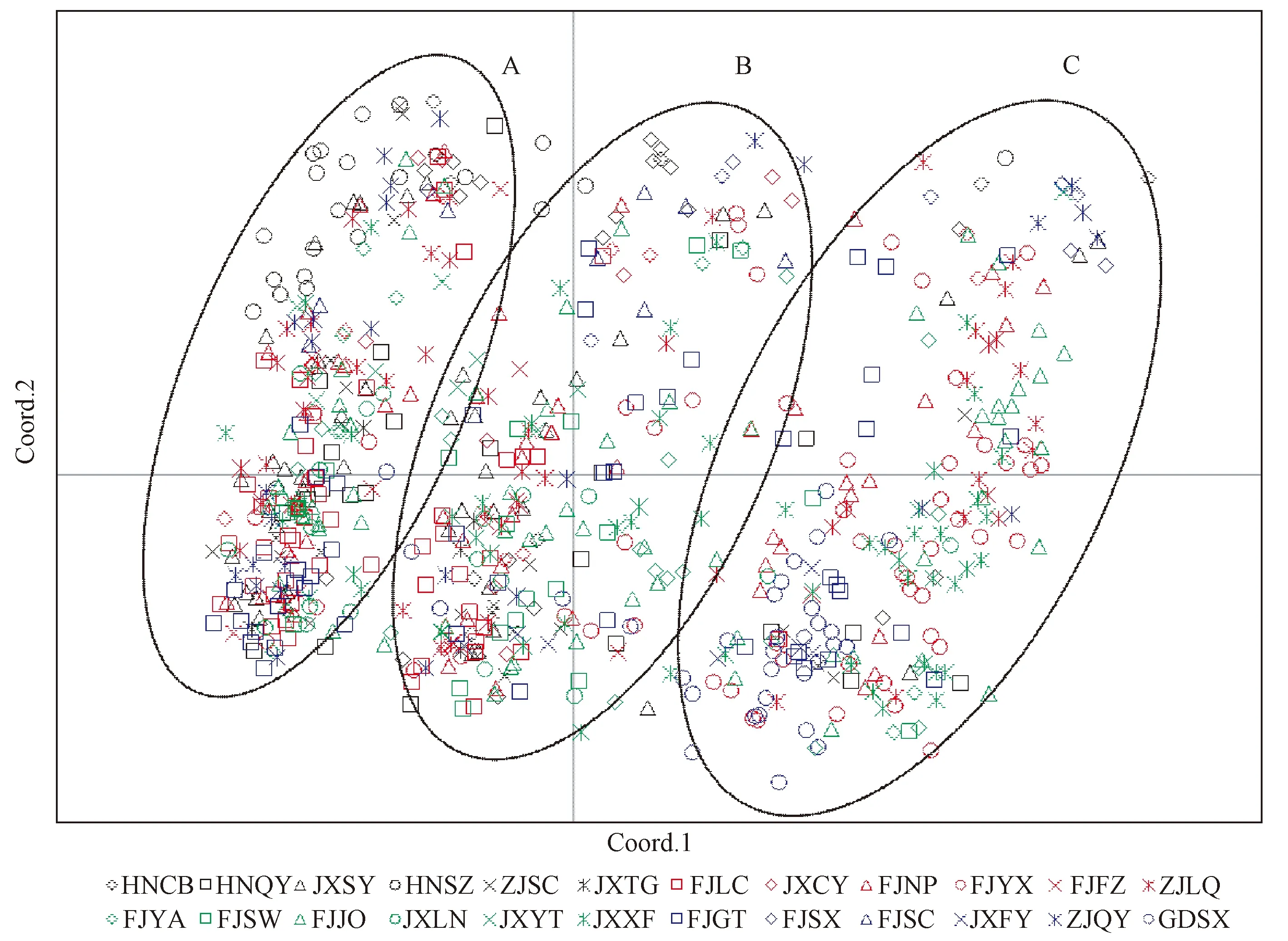
圖4 基于10對SSR引物的734份木荷種質二維PCoA分析Fig.4 Two-dimension principal coordinate analysis of the 734 Schima superba accessions based on 10 SSR primer pairs
為進一步探明種質遺傳多樣性與地理來源的相關性,對24個種質群體繪制基于UPGMA法的樹狀聚類圖(圖5)。24個木荷種質群體間遺傳距離差異很大,介于0.030~0.804之間,平均為0.230。從聚類圖看出,GDSX、HNSZ和FJSX 3個群體與其他群體間遺傳距離均較大,分別單獨聚為一組,其中HNSZ與GDSX間遺傳距離最大,為0.804。HNCB和FJSC聚為一組,它們之間的遺傳距離為0.149。其余19個種質群體全部聚為一組,遺傳距離介于0.030~0.323,其中FJYX與JXXF間的遺傳距離最小,FJSW與JXFY間的遺傳距離最大。
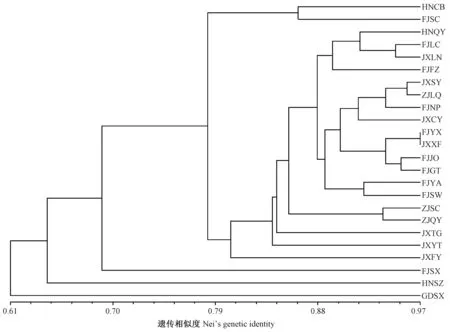
圖5 基于遺傳距離的木荷種質群體聚類Fig.5 Phylogenetic dendrogram based on the genetic distance in the Schima superba germplasm populations
3 討論
無效等位基因(null alleles)會對遺傳學相關研究結果造成顯著影響,如降低群體遺傳多樣性,加大群體間遺傳分化和遺傳距離等,是SSR標記最大的缺陷之一(Paetkauetal., 1997; 文亞峰等, 2013)。本研究中,3對高無效等位基因頻率引物的存在,明顯降低了觀測雜合度(Ho)、期望雜合度(He)和群體總體遺傳多樣性(表2),相應地,使得群體遺傳分化和遺傳距離增大。在進行引物能力評估時,PIC>0.5時,引物貢獻率較高,多態性較好,選取的引物可以最大程度地反映遺傳多樣性(Wangetal., 2014)。本研究中,篩選后的10對SSR引物組合的PIC平均值為0.668,說明選擇的10對SSR引物可以很好反映引物的區分能力,能準確有效地揭示木荷優樹無性系種質的遺傳多樣性。保持林木遺傳多樣性是對林木進一步選擇和改良的前提(李悅等, 2000)。本研究結果顯示,參試木荷優樹無性系種質具有較高的遺傳多樣性,10對多態性SSR引物共檢測出105個等位基因(Na),平均每對引物為10.5個。檢測到等位基因(Na)最多的引物是ss16(16個等位基因),而辛娜娜等(2015)利用ss16引物僅檢測到6個等位基因,Niu等(2012)也只檢測到7個等位基因。本研究中的SSR引物之所以能檢測到更為豐富的變異類型,可能與參試種質豐富、參試種質來源廣泛、群體間的地理位置相距較遠有關。Shannon’s信息指數(I)可以有效反映種質各群體的遺傳多樣性,也是評價遺傳多樣性水平的重要指標(Nybom, 2004)。本研究中木荷優樹無性系種質Shannon’s信息指數(I)為1.473,高于辛娜娜等(2015)利用SSR對木荷一代育種群體遺傳多樣性研究結果(I=1.225)。原因可能有以下2個方面: 一是研究材料來源于木荷主產區的5個省市具有廣泛代表性,這本身就代表著相對較高的遺傳多樣性; 二是與木荷本身的交配系統有關(雌雄同花、蟲媒傳粉且自交不親和)。與其他闊葉樹種相比較,如大花序桉(Eucalyptuscloeziana) (I=1.250)、灰葉胡楊(Populuspruinosa)(I=1.185)等(鄧紫宇, 2012; 張玲等, 2012),本研究中木荷優樹無性系種質表現出較高的遺傳多樣性(I=1.473),這與其本身的生物學特性有關。木荷是一種適應性很強的物種,在群落演替的各個階段都出現,分布廣泛,種群較大,使得木荷在總體上表現出很高的遺傳多樣性。
通過對比分析不同地理來源木荷優樹無性系種質群體間的遺傳多樣性參數,發現FJJO、ZJLQ和JXXF的遺傳多樣性最為豐富,其可能原因為這些地區處于南嶺山脈-武夷山脈附近,與張萍等(2006)劃分的南部種源區和王秀花等(2011)劃分的中心種源區位置大致相同,是木荷的中心產區,其種質不僅最為速生,而且遺傳多樣性也最高。JXFY的遺傳多樣性最低,遺傳變異最小; 這可能與該群體的種質數量較少有關(N=7),如JXTG (N=5)和FJSX(N=9)的遺傳多樣性水平均較低。因此,在今后的木荷種質保存和育種策略制定工作中,應重點考慮遺傳多樣性相對較為豐富的群體(如FJJO、ZJLQ和JXXF等種質群體),優先對這些地區的木荷種質做進一步系統研究和開發利用,同時還應加強對優樹種質的搜集和保存,以拓寬遺傳背景,提高育種成效。育種親本間的遺傳基礎差異越大,產生的雜交后代中越容易選出性狀超越親本和適應性較強的新品種,因此,科學的育種親本選配是雜交育種工作的關鍵,選配親本重要原則之一就是選擇不同生態型、不同地理來源和不同親緣關系的材料作為雜交育種親本(余亞瑩等, 2015)。本研究結果顯示,JXXF與FJYX、JXSY與ZJLQ等種質群體間的遺傳距離較小、親緣關系較近,因此,在木荷雜種育種工作中應避免在這些群體間進行雜交制種; 應在遺傳距離大、親緣關系遠的群體間(如HNCB與FJSX、 GDSX與HNSZ)進行雜交制種,以期獲得更多的遺傳變異和雜種優勢。主坐標分析(PCoA)與STRUCTURE分析屬于2種不同的運算模式,2種分析方法的結合,有助于結果的相互驗證,使結果更為準確,更全面了解不同材料間的遺傳關系(陳斐等, 2013)。在本研究中,主坐標分析(PCoA)及STRUCTURE分析結果均顯示,不同地區種質聚集交錯,與地理來源并無直接關系,這與國內外相關研究結果一致(Laurentinetal., 2006; 辛娜娜等, 2015)。因此,在木荷雜交育種親本選配時不僅要考慮地理遠緣,還應考慮親本群體(個體)間的親緣關系。同時還要根據其他參考性狀,如木材質量、抗逆性等性狀來考慮是否作為雜交育種的親本材料。
分子方差分析(AMOVA)結果表明,木荷優樹無性系種質的遺傳變異主要來源于群體內。因此,在木荷育種工作中,需要側重于優樹個體的選擇和改良,同時兼顧地理群體的選擇。群體遺傳分化的動力主要來源于自然選擇(李康琴, 2013)。木荷優樹無性系種質群體間遺傳分化程度低,原因可能是木荷分布范圍較廣,木荷優樹采集地保留種群一般比較大,呈集群分布,群體間基因流較大而未發生嚴重的遺傳分化。
近年來,基于DNA水平的分子標記在作物關聯作圖分析上得到廣泛應用,而在多年生木本植物中應用相對較少(Zhuetal., 2015; Xuetal., 2015)。對種質遺傳多樣性的研究及遺傳結構的分析,是關聯作圖的前提,當研究所用材料遺傳結構簡單時,關聯分析的功效就會達到最大,結果出現假陽性的可能性也最低(Cardonetal., 2003; Aulchenko, 2011)。在群體遺傳結構分析中,Wang等(2008)在玉米(Zeamays)研究中將Q>0.8的材料認為結構相對單一,在小麥(Triticumaestivum)研究中劉麗華等(2009)將Q>0.6視為結構單一。本文根據種質資源遺傳復雜程度的不同,將Q>0.5作為判斷遺傳結構是否單一的標準,發現71.1%木荷優樹無性系種質遺傳組分相對單一,說明不同地區的木荷優樹無性系種質間存在一定的混雜,同時也表明這些木荷優樹無性系是較好的關聯作圖材料。然而進行表型與基因型的關聯分析時,應具體分析遺傳結構與表型的關系,對關聯分析的結果做出正確的判斷。
4 結論
利用篩選的10對條帶清晰、多態性SSR引物對734份木荷優樹無性系種質進行遺傳多樣性分析,共獲得105個等位基因(Na),多態信息含量(PIC)為0.668。根據地理來源將其劃分為24個種質群體,各種質群體均具有較高的遺傳多樣性。遺傳變異主要存在于群體內,而群體間的交流有限。在選擇高遺傳多樣性群體的基礎上,木荷選育應側重于群體內個體的選擇。主坐標分析(PCoA)及STRUCTURE分析均顯示群體劃分與種質來源不完全相關,在木荷雜交育種親本選配時須同時考慮地理遠緣和親本群體(個體)間的親緣關系。木荷優樹無性系種質的遺傳多樣性豐富,可以為種質創新、優良基因挖掘以及關聯分析提供基礎。
陳 斐,魏臻武,李偉民,等. 2013. 基于SSR標記的苜蓿種質資源遺傳多樣性與群體結構分析. 草地學報, 21(4): 759-768.
(Chen F, Wei Z W, Li W M,etal. 2013. Analysis of genetic diversity and population structure inMedicagogermplasm by SSR markers. Aata Agrestia Sinica, 21(4): 759-768. [in Chinese])
楚秀麗,王 藝,金國慶,等. 2014. 不同生境、初植密度及林齡木荷人工林生長、材性變異及林分分化. 林業科學, 50(6): 152-159.
(Chu X L, Wang Y, Jin G Q,etal. 2014. Variation in growth and wood property and the structure differentiation ofSchimasuperbaplantation with different sites, stand densities and ages. Scientia Silvae Sinicae, 50(6): 152-159. [in Chinese])
鄧紫宇. 2012. 利用SSR分子標記研究大花序桉遺傳結構. 南寧: 廣西大學碩士學位論文.
(Deng Z Y. 2012. Genetic structure ofEucalyptuscloezianaby SSR markers. Nanning: MS thesis of Guangxi University. [in Chinese])
李康琴. 2013. 鵝掌楸屬群體遺傳結構及分子系統地理學分析. 南京:南京林業大學博士學位論文.
(Li K Q. 2013. Studies on populations genetic and molecular phylogeography ofLiriodendron. Nanjing: PhD thesis of Nanjing Forestry University. [in Chinese])
李 悅,張春曉. 2000. 油松育種系統遺傳多樣性研究. 北京林業大學學報, 22(1):12-19.
(Li Y, Zhang C X. 2000. Genetic diversity within a breeding system ofPinustabulaeformis. Journal of Beijing Forestry University, 22(1): 12-19. [in Chinese])
李 培,闕青敏,歐陽昆唏,等. 2016. 不同種源紅椿SRAP標記的遺傳多樣性分析. 林業科學, 52(1): 62-70.
(Li P, Que Q M, Ouyang K X,etal. 2016. Genetic diversity ofToonaciliatafrom different provenances based on sequence-related amplified polymorphism (SRAP) makers. Scientia Silvae Sinicae, 52(1): 62-70. [in Chinese])
劉麗華,王麗新,趙昌平,等. 2009. 光溫敏二系雜交小麥恢復系遺傳多樣性和群體結構分析. 中國生物化學與分子生物學報, 25(9):867-875.
(Liu L H, Wang L X, Zhao C P,etal. 2009. Genetic diversity and alterations of population structure in restorers of dual cross-line hybrid wheat with thermo-photoperiod sensitive male sterile. Chinese Journal of Biochemistry and Molecular Biology, 25(9): 867-875. [in Chinese])
王秀花,陳柳英,馬麗珍,等. 2011. 7年生木荷生長和木材基本密度地理遺傳變異及種源選擇. 林業科學研究, 24(3): 307-313.
(Wang X H, Chen L Y, Ma L Z,etal. 2011. Geographical provenance variation of growth and wood basic density of 7-year-oldSchimasuperbaand its provenance selection. Forest Research, 24(3): 307-313. [in Chinese])
王永康,吳國良,趙愛玲,等. 2014. 棗種質資源的表型遺傳多樣性. 林業科學, 50(10): 33-41.
(Wang Y K, Wu G L, Zhao A L,etal. 2014. Phenotypic genetic diversity of jujube germplasm resources. Scientia Silvae Sinicae, 50(10): 33-41. [in Chinese])
文亞峰,Kentaro Uchiyama,韓文軍,等. 2013. 微衛星標記中的無效等位基因. 生物多樣性, 21(1): 117-126.
(Wen Y F, Kentaro Uchiyama, Han W J,etal. 2013. Null alleles in microsatellite markers. Biodiversity Science, 21(1): 117-126.[in Chinese])
辛娜娜,張 蕊,徐肇友,等. 2015. 木荷1代育種群體遺傳多樣性分析. 林業科學研究, 28(3):332-338.
(Xin N N, Zhang R, Xu Z Y,etal. 2015. Genetic diversity among breeding parents ofSchimasuperbarevealed by SSR. Forest Research, 28(3): 332-338. [in Chinese])
楊愛紅,張金菊,田 華,等. 2014. 鵝掌楸貴州爛木山居群的微衛星遺傳多樣性及空間遺傳結構. 生物多樣性, 22(3): 375-384.
(Yang A H, Zhang J J, Tian H,etal. 2014. Microsatellite genetic diversity and fine-scale spatial genetic structure within a natural stand ofLiriodendronchinense(Magnoliaceae) in Lanmushan, Duyun city, Guizhou province. Biodiversity Science, 22(3): 375-384. [in Chinese])
余亞瑩,邵高能,圣忠華,等. 2015. 國內外香稻資源遺傳多樣性研究. 植物分類與資源學報, 37(6): 871-880.
(Yu Y Y, Shao G N, Sheng Z H,etal. 2015. Genetic diversity of global aromatic rice varieties. Plant Diversity and Resources, 37(6): 871-880. [in Chinese])
張 萍,金國慶,周志春,等. 2004. 木荷苗木性狀的種源變異和地理模式. 林業科學研究, 17(2):192-198.
(Zhang P, Jin G Q, Zhou Z C,etal. 2004. Provenance difference and geographic variation pattern for seedling trait ofSchimasuperba. Forest Research, 17(2):192-198. [in Chinese])
張 萍,周志春,金國慶,等. 2006. 木荷種源遺傳多樣性和種源區初步劃分. 林業科學, 42(2): 38-42.
(Zhang P, Zhou Z C, Jin G Q,etal. 2006. Genetic diversity analysis and provenance zone allocation ofSchimasuperbain China using RAPD markers. Scientia Silvae Sinicae, 42(2): 38-42. [in Chinese])
張 玲,焦培培,李志軍. 2012. 中國新疆灰葉胡楊群體遺傳多樣性的SSR分析. 生態學雜志, 31(11): 2755-2761.
(Zhang L, Jiao P P, Li Z J. 2012. Genetic diversity ofPopuluspruinosapopulations in Xinjiang of China based on SSR analysis. Chinese Journal of Ecology, 31(11): 2755-2761. [in Chinese])
張 蕊,王 藝,金國慶,等. 2013. 施氮對木荷3個種源幼苗根系發育和氮磷效率的影響. 生態學報, 33(12): 3611-3621.
(Zhang R, Wang Y, Jin G Q,etal. 2013. Nitrogen addition affects root growth, phosphorus and nitrogen efficiency of three provenances ofSchimasuperbain barren soil. Acta Ecologica Sinica, 33(12): 3611-3621. [in Chinese])
趙 爽,蘇淑釵,陳志鋼,等. 2016. 基于SSR的遼寧鐵嶺地區平榛遺傳多樣性與居群遺傳結構分析. 果樹學報, 33(1): 24-33.
(Zhao S, Su S C, Chen Z G,etal. 2016. An assessment of the genetic diversity and population genetic structure concerning theCorylusheterophyllaFisch. grown in the Tieling district of Liaoning province, using SSR markers. Journal of Fruit Science, 33(1): 24-33. [in Chinese])
周志春,范輝華,金國慶,等. 2006. 木荷地理遺傳變異和優良種源初選. 林業科學, 19(6): 718-724.
(Zhou Z C, Fan H H, Jin G Q,etal. 2006. Geographic genetic variation and preliminary selection of superior provenance inSchimasuperba. Scientia Silvae Sincae, 19(6): 718-724. [in Chinese])
宗緒曉,關建平,王海飛,等. 2010. 世界栽培豌豆(PisumsativumL.)資源群體結構與遺傳多樣性分析. 中國農業科學, 43(2): 240-251.
(Zong X X, Guan J P, Wang H F,etal. 2010. Population structure and genetic diversity of global pea (PisumsativumL.) germplasm resources. Scientia Agricultura Sinica, 43(2):240-251. [in Chinese])
Aulchenko Y S. 2011. Effects of population structure in genome-wide association studies//Marchini J. Analysis of Complex Disease Association Studies. San Diego: Academic Press.
Cardon L R, Palmer L J. 2003. Population stratification and spurious allelic association. The Lancet, 361(9357): 598-604.
Chuanfu A, Sukumar Saha, Jenkins Johnie N,etal. 2008. Cotton (Gossypiumspp.) R2R3-MYB transcription factors SNP identification, phylogenomic characterization, chromosome location, and linkage mapping. Theoretical and Applied Genetics, 116(7): 1015-1026.
Evanno G, Regnaut S, Goudet J. 2005. Detecting the number of clusters of individuals using the software structure: a simulation study. Molecular Ecology, 14(8): 2611-2620.
Excoffier L, Lischer H. 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10(3):564-567.
Ferr?o L F V, Caixeta E T, Pena G,etal. 2015. New EST-SSR markers ofCoffeaarabica: transferability and application to studies of molecular characterization and genetic mapping. Molecular Breeding, 35(1): 1-5.
Kalinowski S T, Taper M L, Marshall T C. 2007. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Molecular Ecology, 16(5): 1099-1106.
Lassois L, Denancé C, Ravon E,etal. 2016. Genetic diversity, population structure, parentage analysis, and construction of core collection in the French apple germplasm based on SSR markers. Plant Molecular Biology Reporter, 34(4): 827-844.
Laurentin H E, Karlovsky P. 2006. Genetic relationship and diversity in a sesame (SesamumindicumL.) germplasm collection using amplified fragment length polymorphism (AFLP). BMC Genetics, 7(1): 10.
Niu H Y, Li X Y, Ye W H,etal. Isolation and characterization of 36 polymorphic microsatellite markers inSchimasuperba(Theaceae). Journal of Cell Biology, 18(1): 153-166.
Nybom H. 2004. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Molecular Ecology, 13(5): 1143-1155.
Paetkau D, Waits L P, Clarkson P L. 1997. An empirical evaluation of genetic distance statistics using microsatellite data from bear (Ursidae) populations, Genetics, 147(4): 1943-1957.
Peakall R, Smouse P E. 2012. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics, 28(19):2537-2539.
Pritchard J K, Stephens M, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics, 7(4): 574-578.
Rohlf F J. 2000. NTSYS pc, numerical taxonomy and multivariate analysis system, version 2.1. New York, USA: Exeter Publication Ltd Setauket.
Rousset F. 2008. Genepop’ 007: a complete re-implementation of the Genepop, software for windows and Linux. Molecular Ecology Resources, 8(1): 103-106.
Sun G, Bond M, Nass H,etal. 2003. RAPD polymorphisms in spring wheat cultivars and lines with different level ofFusariumresistance. Theoretical and Applied Genetics, 106(6): 1059-1067.
Terzopoulos P J, Bebeli P J. 2008. Genetic diversity analysis of Mediterranean faba bean (ViciafabaL.) with ISSR markers. Field Crops Research, 108(1): 39-44.
Wang R, Yu Y, Zhao J,etal. 2008. Population structure and linkage disequilibrium of a mini core set of maize inbred lines in China. Theoretical and Applied Genetics, 117(7): 1141-1153.
Wang S, Liu Y, Ma L,etal. 2014. Isolation and characterization of microsatellite markers and analysis of genetic diversity in Chinese jujube (ZiziphusjujubaMill.). PloS One, 9(6): e99842.
Xu Y, Huang L, Ji D,etal. 2015. Construction of a dense genetic linkage map and mapping quantitative trait loci for economic traits of a doubled haploid population ofPyropiahaitanensis(Bangiales, Rhodophyta). BMC Plant Biology, 15(1): 228.
Zeka D, Sedlák P, Sedláková V,etal. 2015. Phenotype and molecular diversity evaluation of some wild 2nSolanumspecies (super series Rotata). Chilean Journal of Agricultural Research, 75(2): 147-151.
Zhu Y, Yin Y, Yang K,etal. 2015. Construction of a high-density genetic map using specific length amplified fragment markers and identification of a quantitative trait locus for anthracnose resistance in walnut (JuglansregiaL.). BMC Genomics, 16(1): 614.
(責任編輯 徐 紅)
Analysis of Genetic Diversity inSchimasuperbaPlus Tree Germplasms by SSR Markers
Yang Hanbo1Zhang Rui1Wang Bangshun2Xu Zhaoyou2Chen Huanwei2Zhou Zhichun1
(1. Zhejiang Provincial Key Laboratory of Tree Breeding Research Institute of Subtropical Forestry, Chinese Academy of Forestry Hangzhou 311400; 2. Longquan Forestry Research Institute, Zhejiang Province Longquan 323700)
【Objective】 As a precious broadleaf timber and an efficient tree species for biological fire prevention,Schimasuperbaplays an important role in commercial timber production forests and ecological fireproof forest construction. In depth studies of genetic diversity ofS.superbaplus tree clones using SSR markers are particularly important for conservation, utilization of genetic resources, and future breeding programs for this plant species. 【Method】A total of 734 clones ofS.superbaplus trees from 24 areas of five provinces in China, were analyzed systematically with 10 SSR primer pairs. The GenAIEx 6.5 and CERVUS software were used for genetic diversity parameters calculation, principal coordinates analysis (PCoA) and null alleles detection. NTSYS software was used for cluster analysis based on the matrix of Nei’s genetic identity. The Arlequin software was used for analysis of molecular variance (AMOVA). STRUCTURE 2.3 software was used to analyze genetic structure. 【Result】The results showed that 105 alleles were detected among the germplasm accessions, with an average of 10.5 alleles per pair of primers. The maximum number of alleles was detected in primer ss16 with a value of 16. The Shannon’s information index (I) was ranged from 1.121 to 1.908, with an average of 1.473. The polymorphism information content (PIC) was ranged from 0.557 to 0.807, with an average of 0.668. The expected and observed heterozygosity were 0.713 and 0.735, respectively. The results of principal coordinate analysis (PCoA) and genetic structure analysis were basically consistent with each other, the 734 clones were divided into three groups in PCoA or five subgroups in STRUCTURE analysis. The genetic distance of 24 populations were ranged from 0.030 to 0.804, with an average of 0.230. The results showed that there were close genetic relationship between populations, but, there were still larger genetic distances between some populations, such as HNSZ and GDSX, JXFY and FJSX, etc. The Shannon’s information index (I) of populations were ranged from 0.980 to 1.431, and the genetic diversity was not significantly correlated to geographic distribution. The results of genetic structure analysis indicated that 71.1%S.superbaplus tree clones displayed a simple genetic structure, and the rest 28.9% displayed a mixed genetic structure. The AMOVA results showed that the differentiation among populations contributed to 5.91% of the total genetic variation, and the differentiation within populations contributed 94.09% of the total genetic variation.【Conclusion】 All the results showed that there was a high level of genetic diversity inS.superbaplus tree germplasms, and a significant difference of genetic diversity among populations. When selecting mating parents,the mating pairs should be geographically distant, and genetic relationship between populations or individuals should also be taken into account.
Schimasuperba; plus tree; SSR markers; genetic diversity; genetic structure
10.11707/j.1001-7488.20170506
2016-04-25;
2016-06-15。
“十二五”國家科技支撐計劃課題(2012BAD01B04); 浙江省竹木農業新品種選育重大科技專項竹木育種協作組項目(2012C12908-6); 福建省林木種苗科技攻關五期項目木荷課題; 江西省林業廳林業科技創新專項項目(201503)。
S718.46
A
1001-7488(2017)05-0043-11
*張蕊為通訊作者。
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
