連續流動法測定煙草中總糖、還原糖、氯、鉀含量的不確定度評定
2017-03-23 08:42:17楊乾栩秦云華劉志華U吳億勤蔣次清張承明巍U
食品與機械 2017年12期
許 永 李 超 楊乾栩 - 秦云華 - 蔣 薇 劉志華U - 吳億勤 - 蔣次清 - 張承明 - 劉 巍U
(云南中煙工業有限責任公司技術中心,云南 昆明 650231)
煙草中的糖含量與煙葉品質有著密切的關系,而氯、鉀則與煙草的燃吸性和吸濕性密切相關[1]。煙葉化學成分是評判煙葉品質的重要手段之一[2]。目前在煙草行業中,連續流動法被廣泛用于煙草中總糖、還原糖、氯和鉀含量的快速檢測分析,該方法自動化程度較高。1993年,中國實驗室國家認可委員會公布了《測量不確定度表示指南》[3],自此,不確定度便作為衡量檢測結果可信度的重要指標,陸續應用于化學計量學界,但煙草中對于總糖、還原糖、氯和鉀含量測量不確定度的相關評定分析研究較少[4-8]。目前的研究大多是針對其中的某一種指標來做分析,并計算其擴展不確定度和相應的權重,鮮見有同一種檢測方法同時分析多個指標的不確定度,對不確定度各分量的權重的成因則未見具體分析。為克服前人研究的缺陷,本試驗對連續流動法同時測定煙葉樣品中總糖、還原糖、氯和鉀的檢測過程進行了詳細分析,篩選并明確每一個不確定度的影響因素,同時通過量化各不確定度的分量構成,最終綜合提出該種方法的合成不確定度,以期為實驗室檢測工作的準確性和科學性提供技術支撐。
1 材料與方法
1.1 主要試劑與儀器
樣品:任意抽檢煙葉樣品A,昆明卷煙廠配方庫;
一水葡萄糖:純度≥98%,國藥集團化學試劑有限公司;
氯化鈉:純度≥99.5%,國藥集團化學試劑有限公司;
硫酸鉀:純度≥99%,國藥集團化學試劑有限公司;
其他試劑均為分析純;
連續流動分析儀:AA3型,德國BRAN+LUEBBE公司;
電熱鼓風干燥箱:DHG-9245A型,上海一恒科學儀器有限公司;
電子天平:CP224S型,感量0.000 1 g,德國Sartorius公司;
數字型可調瓶口分液器:50 mL,德國BRAND公司。
1.2 樣品的處理與分析
精確稱取0.250 0 g樣品置于50 mL磨口三角瓶中,加入25 mL冰醋酸,封口后震蕩萃取30 min。用定性濾紙過濾萃取液,棄去前5 mL濾液,收集其余濾液用于分析。按照YC/T31—1996《煙草及煙草制品試樣的制備和水分測定 烘箱法》要求,嚴格進行水分測定,其中天平精度(0.001 g)、烘箱控溫精度[(100±1) ℃]、水分測定值有效期(15 d)和兩次平行測定結果絕對值總差(≤0.10%)都嚴格按標準執行。
依據YC/T 159—2002《煙草及煙草制品水溶性糖的測定 連續流動法》、 YC/T 162—2002《煙草及煙草制品氯的測定 連續流動法》、 YC/T 217—2007《煙草及煙草制品鉀的測定 連續流動法》對樣品進行檢測。具體樣品處理及檢測步驟參見測量流程圖,見圖1。

圖1 測量流程圖Figure 1 Measurement flow chart
1.3 總糖、還原糖、氯、鉀含量
按式(1)計算:
(1)
式中:
P——測定指標的含量,%;
V——萃取液體積,mL;
C——通過標準工作曲線計算得到的樣品成分濃度,g/mL;
W——樣品含水率,%;
m——樣品質量,g。
2 結果與討論
2.1 測量不確定度的評估
2.1.1 不確定度的來源識別 不確定度的來源根據來源因果圖進行識別,見圖2。
通過分析整個測量模型及每一步試驗過程,明確總糖、還原糖、氯和鉀測量的不確定度來源。
2.1.2 水分測量的不確定度u(W) 包括烘箱溫度的不穩定性,含水率測量的重復性,以及分析天平的稱量誤差。
2.1.3 樣品稱量的不確定度u(m) 試驗主要通過分析天平進行樣品稱量,采用的方法為減量法。由于樣品的稱量量較小,可忽略空氣浮力,樣品用量雖小但仍在天平的線性范圍內,因此樣品稱量的不確定度主要是由天平的最大允差造成。
2.1.4 萃取液體積的不確定度u(V) 主要由瓶口分液器刻度和人為讀數誤差造成。試驗過程中,試劑瓶和容量瓶中的萃取液所處的環境溫度差異較小,因此,可以忽略溶劑溫度變化引入的不確定度,具體包括對體積的影響、非水溶液熱膨脹系數差異等。

圖2 不確定度來源的因果圖Figure 2 The causal diagram of the source of uncertainty
2.1.5 測量樣品中總糖、還原糖、氯和鉀濃度的不確定度u(C) 主要來源為標準工作溶液的配制、標準物質定值和非線性工作曲線效應帶來的誤差。
2.1.6 試驗重復性測量的不確定度u(R) 每次外界內部條件不同引入的不確定度。
2.2 測量不確定度分量的量化
2.2.1 樣品水分測量的不確定度u(W)評定 煙草樣品含水率測量的不確定度來源主要有:含水率測量的重復性u(W1),烘箱溫度變化u(W2)和天平稱量不準u(W3)。
(1)u(W1)評定:假設樣品瓶內樣品含水率是均勻的,同一天內同一樣品的6次試驗數據,含水率分別為8.96%,8.89%,8.96%,8.92%,8.87%,8.88%,采用標準偏差計算含水率的重復性。
水分單次試驗的標準偏差為0.040%,平均值標準偏差為:

(2)u(W2)評定:在烘箱溫度[(100±1) ℃]條件下,采用同一樣品進行2次水分測定,每次6個平行樣,數據見表1。假定烘箱溫度為均勻分布,可得溫度對含水率測量影響分量:
(3)u(W3)評定:含水率W按式(2)計算:
(2)
式中:
W——含水率,%;
m0——稱量皿質量,g;
m1——烘前(稱量皿+樣品)質量,g;
m2——烘后(稱量皿+樣品)質量,g。
用同一分析天平進行稱量,假定均勻分布(計算相對不確定度時,采用重復性試驗中數據,m0為16.425 7 g,m1為19.430 6 g,m2為19.161 4 g),具體計算過程見表2。

表1 不同溫度樣品水分含量

表2 u(W3)的計算過程
(4) 合并不確定度:


2.2.3 萃取液體積的不確定度u(V)評定 使用數字型可調瓶口分液器移取萃取液。事先采用衡量法對瓶口分液器進行容量檢定,共做10次測定,其單次試驗的標準偏差為0.080 mL,平均值標準偏差為:
2.2.4 測量煙樣中總糖、還原糖、氯和鉀濃度的不確定度u(C)評定
(1) 由標準物質純度引起的標準溶液定值不確定度u(C0):以生產商提供的信息作為根據,一水葡萄糖純度為98%~100%,擴展不確定度近似為1.0%,按均勻分布計算,其中覆蓋因子k=3,則糖的相對標準不確定度為:
氯化鈉純度為99.5%~100%,擴展不確定度近似為0.25%,按均勻分布計算,其中覆蓋因子k=3,則氯的相對標準不確定度為:
硫酸鉀純度為99%~100%,擴展不確定度近似為0.50%,按均勻分布計算,其中覆蓋因子k=3,則鉀的相對標準不確定度為:
(2) 標準溶液配制引入的標準不確定度u(C1):稱取一水葡萄糖(ρ1)11 g(m1),氯化鈉(ρ2)0.549 8 g(m2),硫酸鉀(ρ3)2.234 0 g(m3),用水稀釋至100 mL,得到儲備液。逐級稀釋儲備液配制得標準工作溶液,見表3。建立標準曲線的過程中,各影響量的不確定度包含在擬合工作曲線的不確定度中,此處不進行單獨計算。
由標準物質稱量引起的相對標準不確定度ur(m):萬分

表3 標準系列溶液配制表
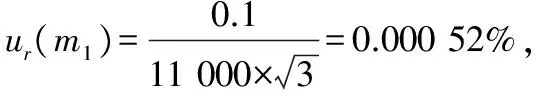

將上述各值代入,計算由標準工作液引入的合成相對標準不確定度為:
ur(C1)氯=0.059%;ur(C1)鉀= 0.058%。
(3) 工作曲線擬合的不確定度u(C2):5個濃度(每個濃度測定3次)的標準工作液由標準物質配制,應用最小二乘法擬合工作曲線,得到回歸方程:
Hj=bCi+a,
(3)
式中:
Hj——第i個標準工作溶液的第j次測定峰高;
b——工作曲線的斜率;
Ci——第i個標準工作溶液的濃度,%;
a——工作曲線的截距。
根據表4、5,得到總糖回歸方程為:Hj=1 438.0Ci+3 366.3。
誤差公式:vi=Hj-(bCi+a),式中vi為殘差。根據總糖回歸方程,可以計算得到理論峰高值與相應的實測峰高值之差,基于Bessel方程,其標準化殘差S為:

表4 總糖標準工作曲線的峰高測定值
(4)
式中:
Hj——第i個標準工作溶液的第j次測定峰高;
b——回歸方程的斜率;
a——工作曲線的截距;
P——樣品檢測次數;
n——標準工作溶液檢測次數;
C——樣品溶液的濃度,%;

i——第i個濃度的標準工作溶液;
j——獲得標準工作曲線的檢測次數。
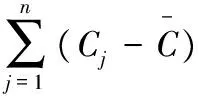
考慮日常檢測平行樣,測試的次數取2次。在計算中,借用重復性測定的數據,試樣溶液雙樣總糖平均值C=18.32%。
同理得濃度為C的還原糖的標準不確定度:

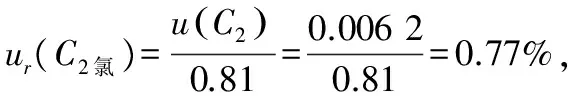

(4) 合并相對標準不確定度:
ur(C)還原糖=1.17%;ur(C)氯=0.78%;ur(C)鉀=1.12%。
2.2.5 試驗重復性引起的不確定度u(R)評定 采用同一樣品,進行6次完整的重復測定,包括稱量樣品、樣品前處理和分析檢測的整個過程,數據見表6。

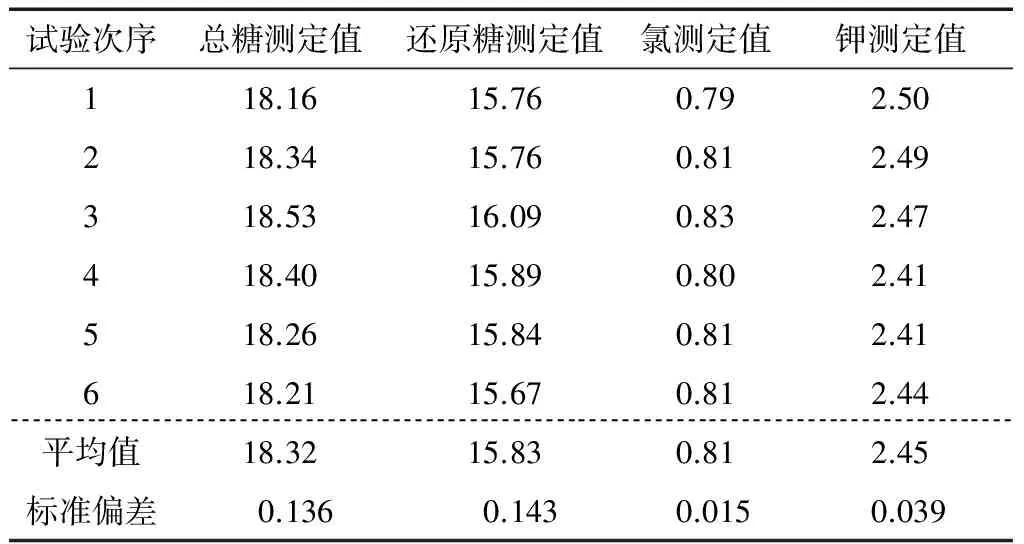
表6 樣品6次測定結果
2.3 計算合成不確定度u(P)
各指標合成相對標準不確定度分別為:
ur(P)總糖=
同理得ur(P)還原糖=1.29%、ur(P)氯=1.16%、ur(P)鉀=1.36%。
根據公式u(P)=P×ur(P)計算各項目的合成不確定度,結果見表7。

表7 各項目合成不確定度
2.4 擴展不確定度分析U(P)及報告
2.4.1 擴展不確定度計算 按正態分布,取95%置信區間,k=2,根據公式U(P)=k×u(P),既得各項目測定結果的擴展不確定度,結果見表8。
2.4.2 測量結果不確定度的表達 按各項目標準進行檢測,樣品中總糖、還原糖、氯、鉀含量測定結果不確定度的表達見表9。

表8 各項目的擴展不確定度

表9 各項目測定結果不確定度的表達
3 結論
采用連續流動法測定煙草中的總糖、還原糖、氯、鉀,分別考慮了水分測定、萃取、過濾、儀器設備以及測定等環節所引入的不確定度分量對測定結果的影響。其測定結果的擴展不確定度分別為0.44%,0.40%,0.02%,0.06%,觀察評定結果,從不同參數的分量大小可以發現,檢測樣品中總糖、還原糖、氯和鉀的濃度引入了最大的不確定度。橫向對比3種待測指標可知,總糖的擴展不確定度最大,還原糖次之,氯最小。這可能與4種物質在煙草中的含量有關,根據4種物質在煙草中的含量均值大小順序:總糖(18.32%)>還原糖(15.83%)>鉀(2.45%)>氯(0.81%)。說明4種指標不確定度大小與其在煙草中的含量呈正比。而最具權重的分量是擬合工作曲線帶來的不確定度。因此,為確保檢測數據可靠準確,在標準工作溶液檢測前,對相關標準溶液的配制和標準曲線制作應嚴格按規定進行。
[1] 李漢超, 王淑嫻. 煙草、煙氣化學及分析[M]. 鄭州: 河南科技出版社, 1991: 28-63.
[2] 邵惠芳, 郭波, 任曉紅, 等. 云南烤煙主產煙區煙葉化學成分比較分析[J]. 安徽農業科學, 2007, 37(7): 1 957-1 959.
[3] ISO. GUM 1995 Guide to expression of uncertainly in measurement, corrected and reprinted [S]. Geneva: International Organization for Standardization, 1995: 1-15.
[4] 中國實驗室國家認可委員會. CNAS-GL06 化學分析中不確定度的評估指南[S]. 北京: 中國計量出版社, 2006: 1-141.
[5] 張婷, 李青誠, 廖曉玲, 等. 氣相色譜法測定煙葉中硫丹殘留量的測量不確定度評價[J]. 計量與測試技術, 2008, 35(2): 27-32.
[6] 李中皓, 唐綱嶺, 陳再根, 等. 頂空-氣質聯用法測定卷煙包裝材料中苯不確定度評價[J]. 質譜學報, 2009, 30(6): 359-363.
[7] 林海濱, 朱永平, 陳星峰, 等. 連續流動法測定煙草中總氮含量的不確定度評定[J]. 福建分析測試, 2009, 18(4): 53-57.
[8] 龐永強, 陳再根, 陳歡, 等. 卷煙側流煙氣中一氧化碳測量不確定度的評定[J]. 現代測量與實驗室管理, 2010(1): 30-31.
猜你喜歡
奧秘(創新大賽)(2023年3期)2023-05-06 01:48:20
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·八年級物理人教版(2019年9期)2019-11-25 07:33:02
當代陜西(2019年8期)2019-05-09 02:22:48
中學生數理化·八年級物理人教版(2019年3期)2019-04-25 06:20:54
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
中學生數理化·八年級物理人教版(2018年3期)2018-05-31 08:52:45
浙江中西醫結合雜志(2017年2期)2017-01-12 18:23:59
當代化工研究(2016年9期)2016-03-20 16:22:08
少兒科學周刊·兒童版(2016年1期)2016-03-14 03:52:21
