鹽酸法舒地爾對大鼠心肌損傷組織中Cx43表達的影響
2016-12-03 06:57:06鄭鵬飛巫相宏黃文馬國添閉奇
山東醫藥 2016年38期
鄭鵬飛,巫相宏,黃文,馬國添,閉奇
(廣西醫科大學第一附屬醫院,南寧530021)
?
·基礎研究·
鹽酸法舒地爾對大鼠心肌損傷組織中Cx43表達的影響
鄭鵬飛,巫相宏,黃文,馬國添,閉奇
(廣西醫科大學第一附屬醫院,南寧530021)
目的 觀察鹽酸法舒地爾(HF)對大鼠心肌損傷組織中縫隙連接蛋白43(Cx43)表達的影響,探討其保護心肌組織的機制。方法 將24只SD大鼠隨機分成空白組、脂多糖(LPS)組、LPS+HF組各8只。LPS+HF組腹腔注射HF 30 mg/kg預處理0.5 h后,尾靜脈注射LPS 1 mg/kg;LPS組腹腔注射HF等量生理鹽水預處理0.5 h后,尾靜脈注射LPS 1 mg/kg;空白組腹腔注射HF等量生理鹽水預處理0.5 h后,尾靜脈注射LPS 等量生理鹽水。尾靜脈注射后6 h處死大鼠,取左心室心肌組織,分別采用Western blot法、熒光定量PCR法檢測心肌組織中的RhoA/ROCK信號通路關鍵蛋白ROCK1、Cx43 蛋白及mRNA。結果 與空白組比較,LPS組ROCK1 mRNA及蛋白表達增加(P均<0.01),Cx43 mRNA及蛋白表達降低(P均<0.01);與LPS組比較,LPS+HF組ROCK1 mRNA及蛋白的表達下降(P均<0.01),Cx43 mRNA及蛋白表達增加(P均<0.01)。結論 鹽酸法舒地爾通過RhoA/ROCK信號通路調節心肌組織中Cx43的表達,從而減輕LPS誘導的心肌損傷。
心肌損傷;脂多糖;法舒地爾;Rho信號通路;ROCK1蛋白;縫隙連接蛋白43;大鼠
細胞縫隙連接是心肌細胞間的電連接形式,調控細胞新陳代謝、內環境穩態、增殖和分化等生理過程。縫隙連接蛋白43(Cx43)是構成心室間隙連接的主要結構蛋白,是心肌細胞間進行小分子量物質通訊和電傳導的基礎,其正常表達和分布是心臟正常電生理活動和協調舒縮的重要保證[1]。已有報道,Cx43參與了急慢性心肌缺血、心力衰竭、心律失常等疾病的發生發展過程[2]。研究指出,RhoA/ROCK通路在心血管疾病的發生發展過程中起關鍵作用[3],ROCK1是該通路的關鍵酶。鹽酸法舒地爾(HF)是惟一種應用在臨床上的Rho激酶抑制劑。已有報道,HF可能阻止和逆轉肺動脈高壓、右心室肥大、心肌細胞損傷的發展[4]。然而,HF改善心肌功能障礙的作用機制尚不清楚。2015年3~12月,本實驗采用脂多糖(LPS)誘導SD大鼠的急性心肌損傷模型來探討HF的治療機制。
1 材料與方法
1.1 材料 SD大鼠,雄性,5周齡,體質量250 g,購自廣西醫科大學動物實驗中心。LPS(美國Sigma公司),HF(山西普德藥業股份有限公司);羊抗兔熒光二抗(美國Licor 公司),兔抗鼠ROCK1抗體(英國Abcam公司),兔抗鼠Cx43抗體及(SAB公司),兔抗鼠GAPDH抗體(Cell Signaling Technology 公司);BCA檢測試劑盒(中國碧云天公司),逆轉錄試劑盒、Taq PCR Master Mix ki(Takara 公司)。
1.2 實驗方法
1.2.1 分組造模與干預處理 24只SD大鼠隨機分成3組:空白組、LPS組、LPS+HF組。LPS+HF組腹腔注射HF 30 mg/kg預處理0.5 h后,尾靜脈注射LPS 1 mg/kg;LPS組腹腔注射HF等量生理鹽水預處理0.5 h后,尾靜脈注射LPS 1 mg/kg;空白組腹腔注射HF等量生理鹽水預處理0.5 h后,尾靜脈注射LPS 等量生理鹽水。
1.2.2 心肌組織取材 尾靜脈注射后6 h,大鼠行腹腔麻醉;取左心室組織,以PBS洗凈,-80 ℃凍存備用。
1.2.3 心肌組織ROCK1、Cx43蛋白檢測 采用Western blot 法。低溫下將心肌組織剪碎,加入RIPA裂解液(含蛋白酶抑制劑 PMSF)快速勻漿,裂解;離心提取組織總蛋白,BCA 法檢測蛋白濃度,100 ℃ 變性 5 min。每孔30 μg蛋白行 SDS-PAGE 電泳,濕轉至PVDF膜,洗膜 3×5 min;5%脫脂奶粉室溫封閉 1 h,洗膜 3×5 min;一抗 4 ℃孵育過夜(ROCK1抗體,1∶1 000;Cx43抗體,1∶500),洗膜 3×5 min;熒光二抗(1∶10 000)室溫下避光孵育 1 h。使用Odyssey雙色紅外激光成像系統掃描,并以目的蛋白與內參 GAPDH灰度比值表示目的蛋白的相對表達水平。
1.2.4 心肌組織ROCK1、Cx43 mRNA檢測 采用實時熒光定量PRC法。TRIzol法提取心肌組織總RNA,分光光度計測定RNA的總濃度。按逆轉錄試劑盒說明書逆轉錄合成cDNA,應用ABI Prism Step One序列檢測系統進行PCR擴增(反應體系20 μL)。95 ℃預變性30 s后進行PCR反應,95 ℃ 5 s、60 ℃ 34 s,共進行40個循環。引物序列:Cx43上游引物5′-GCTCCACTCTCGCCTATGTC-3′,下游引物5′-TAGTTCGCCCAGTTTTGCTC-3′;ROCK1上游引物5′-AAGAGAGTGATATTGAGCAGTTGCG-3′,下游引物5′-TTCCTCTATTTGGTACAGAAAGCCA-3′;GAPDH上游引物5′-GCACCGTCAAGGCTGAGAAC-3′,下游引物5′-CAAAGAGGGTGGAGAGCAAG-3′。用2-ΔΔCt法計算mRNA相對表達量,每個樣品通過管家基因GAPDH作為內參使目標基因標準化。
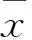
2 結果
2.1 各組心肌組織中ROCK1、Cx43蛋白相對表達量比較 與空白組比較,LPS組中ROCK1蛋白表達顯著上升(P<0.01),Cx43蛋白表達顯著下降(P<0.01);與LPS組比較,LPS+HF組ROCK1蛋白表達顯著下降(P<0.01),Cx43蛋白表達顯著上升(P<0.01)。見表1。
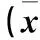
表1 各組心肌組織中ROCK1、Cx43蛋白 相對表達量比較±s)
注:與空白組比較,*P<0.01;與LPS組比較,#P<0.01。
2.2 各組心肌組織中ROCK1、Cx43 mRNA相對表達量比較 與空白組比較,LPS組ROCK1 mRNA表達顯著上升(P<0.01),Cx43 mRNA表達顯著降低(P<0.01);與LPS組比較,LPS+HF組ROCK1 mRNA的表達顯著下降(P<0.01),Cx43 mRNA表達顯著上升(P<0.01)。見表2。
3 討論
近年來,心血管疾病已經成為影響人類健康的重要因素之一,動脈粥樣硬化、心肌梗死及心力衰竭等都能導致心肌功能障礙、心肌收縮不良、心室重構及各種心律失常。大量的實驗和臨床研究指出,RhoA/ROCK通路在心血管疾病的發生發展過程中起關鍵作用。RhoA/ROCK參與了血管平滑肌細胞的收縮、內皮功能障礙、炎癥細胞的聚集和血管重構及心室重構等過程[3]。
表2 各組心肌組織中ROCK1、Cx43 mRNA 相對表達量比較±s)
注:與空白組比較,*P<0.01;與LPS組比較,#P<0.01。
HF是目前惟一一種應用在臨床上的Rho激酶特異性抑制劑,為一種新型異喹啉磺胺衍生物,可以通過與 ATP競爭Rho激酶催化區的 ATP結合位點而阻斷Rho激酶的活性,同時亦可以抑制ROCK1 mRNA及蛋白的表達[5,6]。在臨床上,HF對血管痙攣性心絞痛[7]、穩定型勞力性心絞痛[8]等患者有著顯著的療效。研究指出,Rho激酶活化增加有助于肥胖引起的心臟功能障礙和胰島素抵抗[9]。Rho激酶的表達和活性升高有助于糖尿病心肌病的發展[10];RhoA/ROCK通路有助于缺血性損傷或持續性應激誘導的心肌重塑,從而導致心功能失代償性心力衰竭[11]。HF可以通過抑制ROCK1的表達而明顯改善心肌收縮功能,減少心肌纖維化[12]。目前,Rho激酶通路已經成為治療心血管疾病的一個新靶點。本實驗采用LPS誘導SD大鼠的急性心肌損傷模型來探討HF改善心肌功能障礙的作用機制。結果發現,LPS處理SD大鼠6 h后,其心肌組織中ROCK1的表達水平顯著上升,HF可以降低ROCK1的表達水平,可見,HF通過抑制Rho信號通路而發揮作用。研究指出,在肺靜脈血管內皮細胞中,與Cx43調節主要相關的通路是RhoA/ROCK信號通路而不是PKC信號通路,ROCK1參與對Cx43的調節[13]。而在心肌細胞中,HF是否通過Rho信號通路調節Cx43的表達,這一機制尚不清楚。
縫隙連接介導的心肌細胞間通訊在心肌功能障礙發生發展過程中起關鍵作用,心肌細胞間信息的正常傳遞主要通過縫隙連接來實現。正常心肌細胞Cx43主要表達于相鄰細胞的連接處及閏盤部位。Cx43是心室肌細胞縫隙連接的主要結構蛋白和心肌電生理的主要決定因素,是心肌細胞間進行小分子量物質通訊和電傳導的基礎。其表達正常和分布是心臟正常電活動維持和舒縮功能協調的必要條件[1]。研究指出,破壞心肌細胞間正常分布的Cx43,能增加室性心律失常和猝死的發生[14];折返性心律失常及慢性肥厚心臟傳導異常,與Cx43的表達降低有關[15];心室肌細胞中Cx43的下調和重新分布,有助于心室結構重塑及左心室纖維化的發生[16];心肌缺血或者慢性肥厚的心室肌細胞中,Cx43表達降低[17]。缺氧可下調乳鼠心肌細胞Cx43表達量,導致心肌細胞縫隙連接重構,誘發惡性心律失常[18]。研究指出,LPS損傷心肌細胞后,心肌細胞中的Cx43表達會顯著降低,從而影響心肌細胞正常功能的發揮[19]。本實驗發現,在LPS作用SD大鼠6 h后,心肌組織中Cx43的表達顯著降低,而HF預處理0.5 h可以干預上述結果。因此,我們認為HF通過Rho信號通路調節損傷的心肌組織中Cx43表達,從而改善心肌功能障礙。
[1] Schulz R, G?rge PM, G?rbe A, et al. Connexin 43 is an emerging therapeutic target in ischemia/reperfusion injury,cardioprotection and neuroprotection[J]. Pharmacol Ther, 2015,153(2):90-106.
[2] Wu W, Li Y, Lu Z, et al. Increased susceptibility to ischemia-induced ventricular tachyarrhythmias in depressed rats: Involvement of reduction of connexin 43[J]. Exp Ther Med, 2012,3(2):192-194.
[3] Surma M, Wei L, Shi J, et al. Rho kinase as a therapeutic target in cardiovascular disease[J]. Future Cardiol, 2011,7(5):657-671.
[4] Sun XZ, Li SY, Tian XY, et al. Effect of fasudil on hypoxic pulmonary hypertension and right ventricular hypertrophy in rats[J]. Int J Clin Exp Pathol, 2015,8(8):9517-9521.
[5] Shi J, Lei W. Rho kinases in cardiovascular physiology and pathophysiology: the effect of fasudil[J]. J Cardiovasc Pharmacol, 2013,62(4):341-354.
[6] Zhang J, Bian HJ, Li XX, et al. ERK-MAPK signaling opposes Rho-kinase to reduce cardiomyocyte apoptosis in Heart ischemic preconditioning[J]. Mol Med, 2010,16(7-8):307-315.
[7] Masumoto A, Mohri M, Shimokawa H, et al. Suppression of coronary artery spasm by the Rho-kinase inhibitor fasudil in patients with vasospastic angina[J].Circulation, 2002,105(13):1545-1547.
[8] Fukumoto Y, Mohri M, Inokuchi K, et al. Anti-ischemic effects of fasudil,a specific Rho-kinase inhibitor,in patients with stable effort angina[J]. J Cardiovasc Pharmacol, 2007,49(3):117-121.
[9] Soliman H, Nyamandi V, Garcia-Patino M, et al. Partial deletion of ROCK2 protects mice from high-fat diet-induced cardiac insulin resistance and contractile dysfunction[J]. Am J Physiol Heart Circ Physiol, 2015,309(1):70-81.
[10] Waddingham MT, Edgley AJ, Astolfo A, et al. Chronic Rho-kinase inhibition improves left ventricular contractile dysfunction in early type-1 diabetes by increasing myosin cross-bridge extension[J]. Cardiovasc Diabetol, 2015,22(14):92-105.
[11] Wang N, Guan P, Zhang JP, et al. Fasudil hydrochloride hydrate, a Rho-kinase inhibitor, suppresses isoproterenol-induced heart failure in rats via JNK and ERK1/2 pathways[J]. J Cell Biochem, 2011,112(7):1920-1929.
[12] Mera C, Godoy I, Ramírez R, et al. Mechanisms of favorable effects of Rho kinase inhibition on myocardial remodeling and systolic function after experimental myocardial infarction in the rat[J]. Ther Adv Cardiovasc Dis, 2016,10(1):4-20.
[13] Zhang J, Yang GM, Zhu Y, et al. Role of connexin 43 in vascular hyperpermeability and the relationship to the Rock 1-MLC20 pathway in septic rats[J]. Am J Physiol Lung Cell Mol Physiol, 2015,309(11):1323-1332.
[14] Maruyama D, Hirata N, Tokinaga Y, et al. Nitrite reduces ischemia-induced ventricular arrhythmias by attenuating connexin 43 dephosphorylation in rats[J]. Anesth Analg, 2016,122(2):410-417 .
[15] Formigli L, Ibba-Manneschi L, Perna AM, et al. Altered Cx43 expression during myocardial adaptation to acute and chronic volume overloading[J]. Histol Histopatho, 2003,18(2):359-369.
[16] Quang KL, Maguy A, Qi XY, et al. Loss of cardiomyocyte integrin-linked kinase produces an arrhythmogenic cardiomyopathy in mice[J]. Circ Arrhythm Electrophysiol, 2015,8(4):921-932.
[17] Chanson M, Derouette JP, Roth I, et al. Gap junctional communication in tissue inflammation and repair[J]. Biochim Biophys Acta, 2005,1711(2):197-207.
[18] Zeevi-Levin N, Barac YD, Reisner Y, et al. Gap junctional remodeling by hypoxia in cultured neonatal rat ventricular myocytes[J]. Cardiovasc Res, 2005,66(1):64-73.
[19] Fernandez-Cobo M, Gingalewski C, Drujan D, et al. Downregulation of connexin 43 gene expression in rat heart during inflammation.The role of tumour necrosis factor[J]. Cytokine, 1999,11(3):216-224.
國家自然科學基金資助項目(81360057)。
巫相宏(E-mail: whw780@126.com)
10.3969/j.issn.1002-266X.2016.38.008
R966
A
1002-266X(2016)38-0026-03
2016-04-23)
