以不典型大腦病變起病的視神經脊髓炎譜系疾病1例報告
2016-09-19 09:34:49宋洋洋韓金鳴曹青青
中風與神經疾病雜志 2016年7期
關鍵詞:信號
宋洋洋,韓金鳴,曹青青,金 濤
?
以不典型大腦病變起病的視神經脊髓炎譜系疾病1例報告
宋洋洋,韓金鳴,曹青青,金濤
2015年6月發表于美國神經病學會 Neurology雜志上的視神經脊髓炎譜系疾病(neuromyelitis optica spectrum disorders,NMOSD) 新標準分為 AQP4 抗體陽性 NMOSD 和 AQP4 抗體陰性 NMOSD。以不典型大腦病變起病的NMOSD罕見報道,現將我院收治的1例病例報道如下。
1 臨床資料
患者,女,62歲,因間斷失認、右眼視力下降2 y余,雙下肢無力伴尿便潴留6 m,加重20 d于2015年7月10日入院。患者于入院前2y余無明顯誘因出現失認、右眼視力下降,就診于多家醫院,頭部MRI(2013年3月,吉林大學中日聯誼醫院)示:左側枕葉、胼胝體、側腦室旁異常信號及強化信號(見圖1);頭部MRS(2013年3月,復旦大學附屬華山醫院)示:左頂枕葉側腦室后角區域病灶內細胞增殖代謝升高,Cho峰增高,NAA峰降低,Cho/NAA為1.334~1.855左右(見圖1);右枕腦室旁行穿刺活檢(2013年4月,復旦大學附屬華山醫院)示:少量腦組織內髓鞘斷裂缺失,大量泡沫細胞形成,符合脫髓鞘性病變。入院前6 m無明顯誘因出現雙下肢無力,左側明顯,走路左偏,家屬攙扶可行走,伴針刺樣疼痛,呈放射狀,服用“鎮痛藥”后疼痛略緩解。入院前20 d上述癥狀加重,表現為不能行走,雙下肢持續疼痛,尿便潴留加重,為求進一步診治入我科。病程中無疫苗接種史、發熱、腹瀉,飲食睡眠可,體重無明顯變化。既往:高血壓病史20 y,血壓最高200/120 mmHg,規律服用“苯磺酸左旋氨氯地平,倍他樂克”,收縮壓控制在140~150 mmHg。入院查體:體溫36.4 ℃,血壓145/88 mmHg,心率75次/分,內科系統查體未見異常。神清語明,視力視野概測正常,雙側瞳孔等大同圓,直徑約3.0mm,直間接對光反射靈敏,腦神經未見異常。雙上肢肌力5級,雙下肢近端肌力5級,右下肢遠端肌力4級,左下肢遠端肌力0級,左足背屈不能。四肢肌張力正常。雙上肢腱反射正常,雙下肢腱反射活躍。右側Babinski征及右側Chaddock 征(+)。T8以下痛覺減退,關節位置覺障礙,無項強,Kernig征陰性。輔助檢查:(2013年4月,上海長海醫院)腦脊液無色透明,常規生化大致正常,24 h IgG鞘合成率4.58 mg/dl(參考值9.9~3.3 mg/dl),一般細菌、真菌培養、墨汁染色、結核菌涂片(-)。血沉、抗核抗體測定(ANA)、抗核提取物抗體測定、抗平滑肌抗體測定、抗雙鏈DNA測定、抗心磷脂抗體、抗中性粒細胞胞漿抗體、抗組蛋白抗體(AHA)測定、抗單鏈DNA測定、抗核糖體抗體測定、抗核小體抗體測定、抗腎小球基底膜抗體測定、抗線粒體抗體測定、抗生殖細胞核抗原抗體、抗中性粒細胞胞漿抗體、類風濕因子RF測定大致正常。入院后輔助檢查:腦脊液無色透明,壓力130 mmH2O,常規生化:潘氏反應+,蛋白1.78 g/L(參考值0.15~0.45 g/L),白細胞36×106,糖、氯大致正常,IgG合成指數 68 mg/24 h(參考值<7.0 mg/1 mg/24 h),血清、腦脊液抗水通道蛋白(AQP4)抗體陽性()。胸腰椎MRI(2015年7月)示:胸10至腰1椎體水平脊髓內異常信號(見圖2)。綜上,臨床診斷為視神經脊髓炎譜系疾病、高血壓病3級(極高危險組)。給予甲強龍1000 mg沖擊治療12 d后患者尿便潴留較前好轉,改為強的松 60 mg口服后出院。復查胸腰段MRI:胸10-11間盤水平至腰3椎體水平脊髓內改變,較前片病灶面積略有減小。
2 討 論
NMOSD 概念最早是由 Wingerchuk于2007年提出[1]。2015年Neurology上更新了NMOSD的診斷標準[2]:(1)AQP4-IgG陽性NMOSD診斷標準:①至少1項核心臨床特征;②用可靠的方法檢測AQP4-IgG陽性(推薦CBA法);③排除其他診斷。核心臨床特征:視神經炎、急性脊髓炎、最后區綜合征,無其他原因能解釋的發作性呃逆、惡心、嘔吐、其他腦干綜合征、癥狀性發作性睡病、間腦綜合征,腦MRI有NMOSD型間腦病變、大腦綜合征伴有NMOSD型大腦病變。(2)AQP4-IgG陰性或未檢測的NMOSD診斷標準:①至少2項核心臨床特征,其中至少1項為視神經炎、急性長脊髓炎或延髓最后區綜合征;② 空間多發(2項或以上不同的臨床核心特征);③滿足MRI附加條件。AQP4-IgG陰性或未檢測的NMOSD的MRI附加條件:(1)急性視神經炎,需腦MRI為下列之一表現:①腦MRI正常或僅有非特異性白質病變;②視神經MRI長T2信號或增強的T1信號>1/2視神經長度,或病變累及視交叉。(2)急性脊髓炎,MRI長脊髓病變 3連續椎體節段,或有脊髓炎病史的患者MRI示相應脊髓萎縮3連續椎體節段。(3)最后區綜合征:延髓背側/最后區病變(4)急性腦干綜合征:腦干室管膜周圍病變。
部分NMOSD患者早期影像學表現不典型,其中,早期以腦部病變起病者較為少見。多數NMOSD患者腦部影像學改變與大腦的 AQP4 高表達區域具有相當的一致性,腦部病灶主要分布在三、四腦室周圍、導水管、延髓中央管周圍及下丘腦。僅有少數侵犯腦實質,絕大多數病灶為非特異性改變,多為非典型的皮質下或毗鄰腦室的異常信號[3]。
國內李珍珍等[4]報道1例視神經脊髓炎廣泛大腦半球病灶,其為43歲女性,頭部MRI示病灶位于皮髓質交界區以及深部白質,與本病類似。國外Tahara 等[5]報道了日本3例NMOSD患者腦病樣癥狀,MRI示腦皮質異常;Kim 等[6]回顧性分析韓國215例NMOSD患者,指出大腦皮質病灶可能與炎性細胞通過受損的血腦屏障進入相鄰的皮質有關。
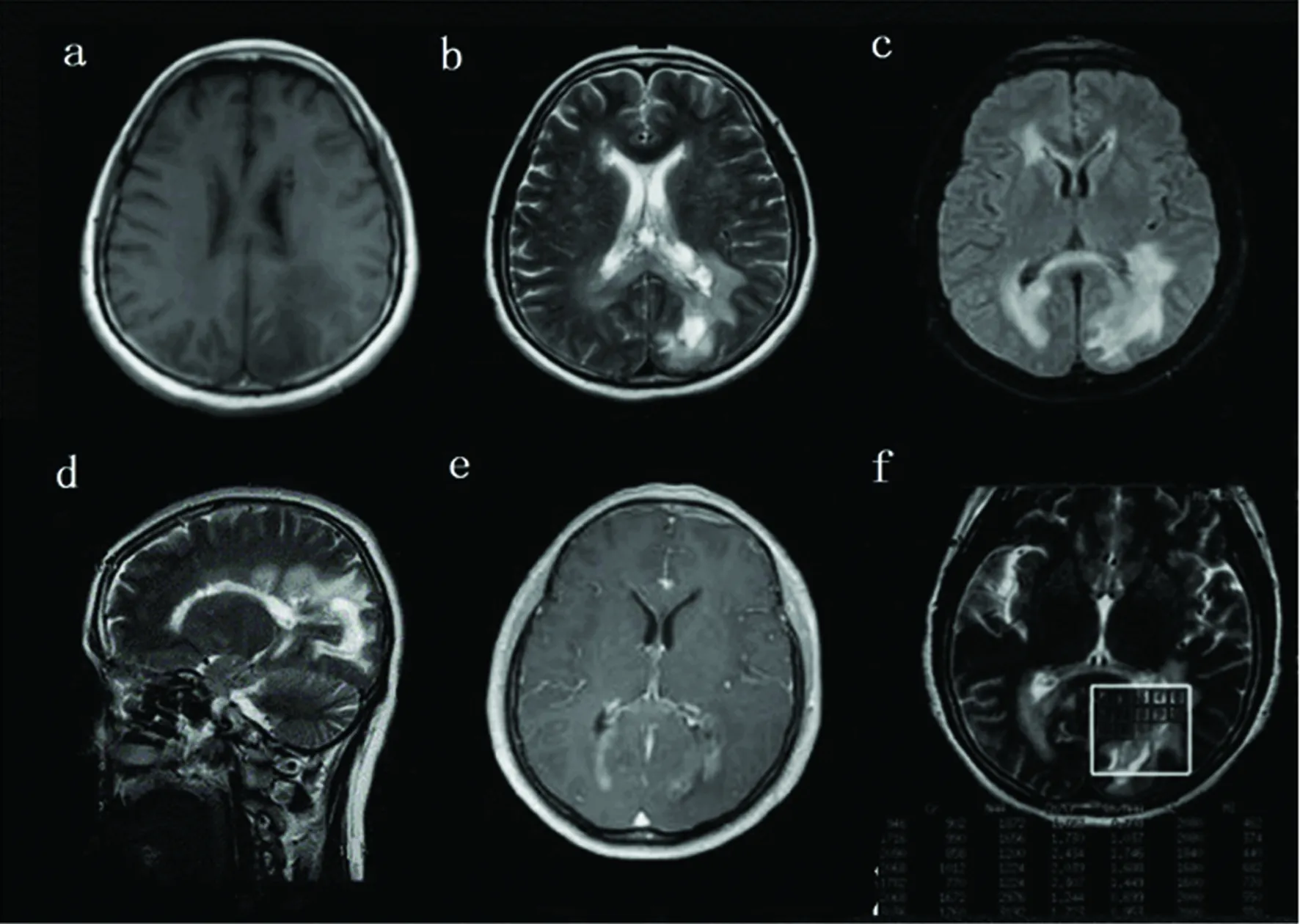
圖1 頭部MRI:左側枕葉、胼胝體及雙側側腦室旁異常信號。T1WI(a)呈稍低信號,T2WI(b)呈稍高信號,Flair(c)呈稍高信號,矢狀T2(d)呈稍高信號,增強(e)可見強化影。頭部MRS(f):Cho峰增高,NAA峰降低,Cho/NAA為1.334~1.855左右

圖2 胸椎MRI:胸10至腰1椎體水平脊髓內異常信號。矢狀位T2 WI(a)呈高信號,軸位T2WI(b)呈高信號
本例患者首次發病頭部MRI顯示腦內多發病灶,且呈大面積融合病灶,應與急性播散性腦脊髓炎(acute disseminated encephalomyelitis,ADEM)鑒別。ADEM呈單相病程,兒童多見,常繼發于感染、出疹及疫苗接種后。本例患者為老年女性,病前無感染及疫苗接種史,隨疾病進展,出現2次雙下肢無力伴尿便障礙,胸腰段MRI脊髓內可見長節段橫貫性異常信號,血清、腦脊液抗水通道蛋白抗體陽性(),結合患者癥狀、體征及輔助檢查,符合2015年Neurology關于NMOSD的診斷標準中AQP4抗體陽性的NMOSD診斷標準。
因此,以不典型大腦病變起病的患者應警惕NMOSD的可能,如條件允許應進一步完善視覺誘發電位、脊髓核磁、血清及腦脊液AQP4 抗體檢測,并做好疾病的長期隨訪,對進一步明確診治十分必要。
[1]Wingerchuk DM,Lennon VA,Lucchinetti CF,et al. The spectrum of neuromyelitis optica[J]. Lancet Neurol,2007,6(9):805-815.
[2]Wingerchuk M,Banwell B,Bennett JL,et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders[J]. Neurology,2015,85(2):177-189.
[3]賈建平. 中樞神經系統脫髓鞘疾病[A]. 吳江. 神經病學[M]. 第2版. 北京:人民衛生出版社,2010. 246-249.
[4]李珍珍,任士卿. 視神經脊髓炎廣泛大腦半球病灶1例[J]. 臨床薈萃,2011,26(20):1815.
[5]Tahara M,Ito R,Tanaka K,et al. Cortical and leptomeningeal involvement in three cases of neuromyelitis optica[J]. Eur Neurol,2012,19:e47-48.
[6]Kim W,Lee JE,Kim SH,et al. Cerebral cortex involvement in neuromyelitis optica spectrum disorder[J]. Clin Neurol,2016,12:188-193.
1003-2754(2016)07-0653-02
R744.3
2016-05-06;
2016-07-12
(吉林大學白求恩第一醫院神經內科,吉林 長春 130021)
金濤,E-mail:drtao. jin@hotmail.com
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
