氯諾昔康干混懸劑的制備及其質量考察
2016-07-27 00:54:39沙崢張洪彭銳張英魏丹蕓
廣東藥科大學學報 2016年3期
沙崢,張洪,彭銳,張英,魏丹蕓
?
氯諾昔康干混懸劑的制備及其質量考察
沙崢,張洪,彭銳,張英,魏丹蕓
(武漢大學人民醫院 藥學部,湖北武漢430060)
摘要:目的制備氯諾昔康干混懸劑并對其質量進行考察。方法通過單因素試驗,考察不同輔料對氯諾昔康干混懸劑沉降體積比及再分散性的影響,再運用正交試驗優選出最優處方工藝;采用高效液相色譜法測定干混懸劑中氯諾昔康的質量分數,并對其溶出度及穩定性進行考察。結果最佳處方工藝為:以質量分數8%的CMC-Na和5%的黃原膠共同作為助懸劑,15%的微晶纖維素為崩解劑,15%的聚乙烯吡咯烷酮K30為黏合劑;3批樣品標示質量分數平均值為98.7%;pH 7.4磷酸鹽緩沖液作為溶出介質時,氯諾昔康溶出速度快且較平緩;穩定性試驗顯示制劑的各項檢測指標均無明顯變化。結論優化后的處方工藝簡單、可行、穩定性可控、重復性好,所制備的制劑符合干混懸劑質量要求。
關鍵詞:氯諾昔康;干混懸劑;處方工藝;高效液相色譜法;穩定性
氯諾昔康是一種新型非甾體抗感染藥,常用于治療手術鎮痛、急慢性骨關節炎、脊椎炎和風濕性關節炎等。由于氯諾昔康血漿半衰期短,不良反應較輕,耐受性好,抗感染鎮痛效果顯著,從而受到越來越多醫生和患者青睞[1]。目前,國內外上市的氯諾昔康有片劑和針劑2種。口服片劑易吸收,但生物利用度低,且受飲食影響易導致吸收慢[2]。針劑的不良反應主要是惡心、嘔吐、胃痛、腹瀉等胃腸道反應,發生率為10%,高于片劑的5.45%[3]。口服干混懸劑是將藥物用適宜方法制成粉末狀或顆粒狀的制劑,使用時加水能夠迅速分散成混懸劑[4],解決了混懸劑在儲存過程中的穩定性問題。藥物在混懸劑
網絡出版時間:2016-05-25 10:17 網絡出版地址:http://www.cnki.net/kcms/detail/44.1413.R.20160525.1017.001.html中以微粒狀態分散,分散度大,對胃腸道的刺激小,能被胃腸道迅速吸收,有利于提高生物利用度[5],可進一步提高患者對藥物劑型的順應性,改善病人的服藥順應性。本文通過對干混懸劑輔料的考察,采用正交試驗設計篩選氯諾昔康干混懸劑的處方,以HPLC法對氯諾昔康的質量分數及溶出度進行測定,并通過影響因素試驗及加速試驗考察其穩定性,為氯諾昔康的臨床應用提供依據。
1 儀器與試藥
1.1 儀器
BT214D分析電子天平(德國賽多利斯公司);WD-2A藥物穩定性檢查儀(天津精拓儀器科技有限公司);YNG-9245A高溫干燥箱(上海姚氏儀器設備廠);DK-24S電熱恒溫水浴鍋(艾斯派克公司);Agilent 1100型高效液相色譜儀(美國 Agilent公司);ZRS-8G型智能溶出試驗儀(天津市天大天發科技有限公司)。
1.2 試藥
氯諾昔康原料藥(武漢市東康源科技有限公司,批號20140125,質量分數>99.8%);乳糖(武漢市長福源醫藥輔料有限公司,批號20140309);羧甲基纖維素鈉(CMC-Na,濟南天勝化工有限公司,批號20150103);黃原膠(上海恒生化工有限公司,批號20141108);滑石粉(宜興凱利達化學有限公司,批號20150201);聚乙烯吡咯烷酮K30(PVP K30,大連美侖生物技術有限公司,批號20141112);醋酸鈉(常熟南湖化工有限責任公司);甲醇(色譜純,Tedia company);羥丙基甲基纖維素 (HPMCK15M和HPMCK100M)、乙醇、微晶纖維素(MCC)均為國藥集團化學試劑有限公司提供;水為實驗室自制純化水。
2 方法與結果
2.1 原輔料相容性試驗
以乳糖、MCC、各型號的 HPMC、黃原膠、CMC-Na作為輔料,考察原輔料之間的相容性。按照100∶200∶1∶1∶1∶1∶1(質量比)稱取氯諾昔康、乳糖、MCC、HPMCK15M、HPMCK100M、黃原膠、CMC-Na,分別過80目篩后混合均勻。將混合物分別在高濕(RH75%)、強光[(4 500±500)lx]和高溫(60℃)的條件下放置10 d,考察原料藥與原輔料混粉的性狀、干燥失重及含量的變化情況。結果表明,濕度對原料藥及原輔料混粉有一定的影響,高溫跟強光則沒有顯著影響。因此,在制備氯諾昔康干混懸劑時應
控制環境濕度,注意防潮。
2.2 填充劑、黏合劑、崩解劑的選擇
通過預試驗,選擇乳糖、10%(質量分數,下同)PVP(溶于體積分數50%乙醇溶液中)和MCC分別作為填充劑、黏合劑和崩解劑,制備得到的干混懸劑外觀美觀,不易吸濕,性質穩定,分散性較好,溶出度在規定范圍內。
2.3 助懸劑種類的選擇
通過等量遞加法分別將黃原膠、HPMCK15M、HPMCK100M、CMC-Na和混粉(黃原膠+CMC-Na)5種助懸劑各1.3 g,加入到5份處方中(處方中其他成分的量固定:氯諾昔康0.32 g,乳糖6.88 g,MCC 1.5 g,滑石粉0.1 g),混合均勻,依法制備干混懸劑。依據《中國藥典》[6]附錄混懸劑項下的要求,對沉降體積比及再分散性進行考察:將適量所制備的5種干混懸劑分別置于50 mL具塞量筒中,加水至刻度,密塞,用力振搖1 min,觀察溶解及分散情況,并記下此時混懸物的初始高度(H0),然后靜置3 h,記下此時混懸物的最終高度(H),沉降體積比=H/H0;將混懸液繼續沉降1 d,將具塞量筒倒轉180°,倒轉1次停留5 s,記為振搖1次。重復上述振搖過程,直至干混懸劑再次均勻分散,記下混懸液振搖的次數。振搖次數越少,說明該干混懸劑的分散性越好。
分別記錄各處方中干混懸劑的沉淀狀況、溶解情況、沉降體積比及再分散性,以無沉淀、易溶解、沉降體積比>0.9、再分散性好作為篩選助懸劑的標準,結果見表1。可見,黃原膠和CMC-Na的混粉作為助懸劑時,所制備的干混懸劑無沉淀產生,溶解性好,沉降體積比符合要求,再分散性好。

表1 不同助懸劑的考察結果Table 1 The result of the different suspending agent
2.4 正交試驗優化氯諾昔康干混懸劑的處方
通過對單因素試驗的考察,確定MCC的用量、黃原膠的用量、CMC-Na的用量、PVP K30的質量濃度作為影響干混懸劑的4種主要因素,每個因素設置3個水平,以沉降體積比為考察指標,采用L9(34)正交試驗篩選最優處方。因素與水平見表2,正交試驗結果見表3,方差分析結果見表4。
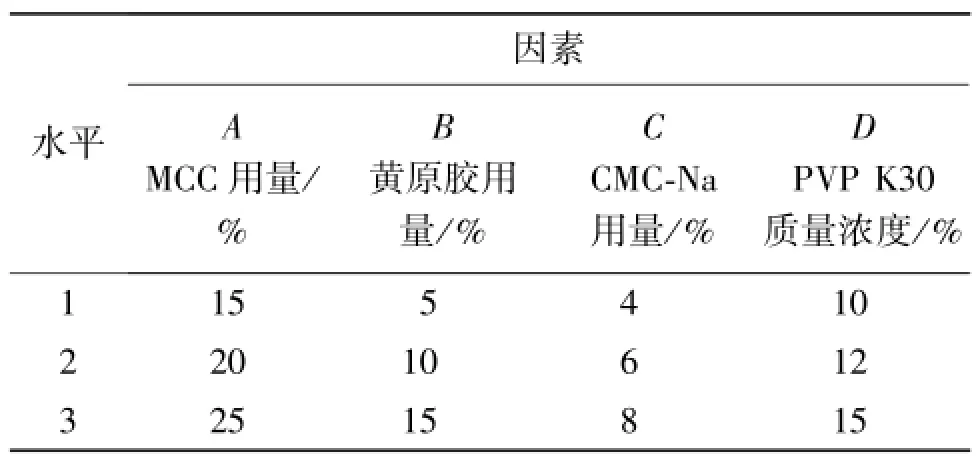
表2 正交試驗因素水平表Table 2 The factor and levels of orthogonal test
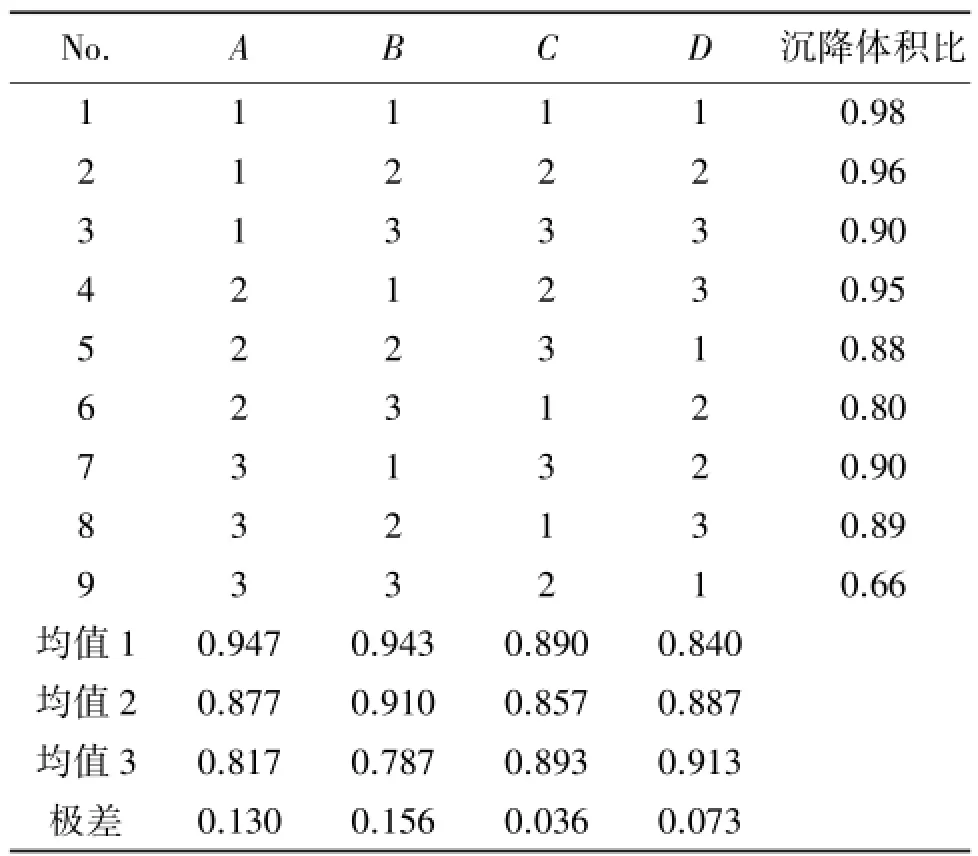
表3 正交試驗設計與結果Table 3 The results of orthogonal test

表4 正交試驗結果方差分析表Table 4 Analysis of variance
可見,各因素對沉降體積比的影響大小順序為B>A>D>C,各因素水平的優劣順序分別為:A因素1>2>3,B因素1>2>3,C因素3>1>2,D因素3>2>1。方差分析結果表明,4個因素對助懸劑沉降體積比的影響均無統計學意義。根據以上結果,確定A1B1C3D3為該干混懸劑的最佳處方,即MCC用量為15%,黃原膠用量為5%,CMC-Na用量為8%,PVP質量濃度為15%。
2.5 驗證試驗
按正交試驗優化的處方,稱取處方量的氟諾昔康及輔料,粉碎后分別過80目篩,按等量遞加稀釋法將藥物與輔料混合均勻,加入適量10%PVP(50%乙醇溶液)的黏合劑制軟材,過25目篩制粒,在60℃烘箱中干燥1 h,過20目篩整粒,再向干燥顆粒中加入適量滑石粉,混勻,即得氯諾昔康干混懸劑。同法制備 3批樣品 (批號:2015061502、2015061506、2015061508),分別測定沉降體積比,結果測得3批樣品的沉降體積比分別為0.98、0.99、0.98,均符合要求,表明正交試驗優化的處方工藝的重復性較好。
2.6 氯諾昔康干混懸劑的質量分數測定
2.6.1 色譜條件 色譜柱:Diamonmsil C18(4.6 mm× 200 mm,5 μm);流動相:pH 5.8的0.05 mol/L醋酸鈉-甲醇(體積比10∶90);流速:1.0 mL/min;檢測波長:384 nm;柱溫:30℃;進樣量:20 μL。
2.6.2 對照品溶液的制備 精密稱取氯諾昔康對照品10 mg,置于50 mL量瓶中,用流動相溶解并稀釋至刻度,精密量取4 mL置于10 mL量瓶中,用流動相溶解并稀釋至刻度,搖勻,即得。
2.6.3 供試品溶液的制備 精密稱取本品適量(批號:2015061502,相當于氯諾昔康約 8 mg),置于100 mL容量瓶中,加適量流動相,超聲(溫度控制在60℃)振搖使溶解,用流動相稀釋至刻度,并用0.45 μm微孔濾膜濾過,取續濾液,即得。
2.6.4 陰性對照品溶液的制備 根據處方制備不含氯諾昔康的樣品,按“2.6.3”項下制備供試品溶液的方法制備陰性對照品溶液。
2.6.5 專屬性試驗 分別取供試品、對照品、陰性對照品溶液各5 mL,置于10 mL容量瓶中,用流動相稀釋至刻度,搖勻,分別精密量取20 μL,注入液相色譜儀中進行測定,結果見圖1。可見,氯諾昔康的保留時間為7.9 min,陰性對照品在氯諾昔康對照品色譜峰相應的保留時間處沒有峰出現,表明輔料對供試品中氯諾昔康的測定無干擾。
2.6.6 線性關系考察 分別量取氯諾昔康對照品溶液2、5、10、15、20、25 mL,置于25 mL容量瓶中,加流動相稀釋至刻度,搖勻,用0.45 μm微孔濾膜濾過,分別精密吸取上述對照品溶液20 μL,注入液相色譜儀中,以質量濃度為橫坐標,峰面積為縱坐標進行線性回歸,得回歸方程A=29.562ρ+496.32(r= 0.999 7),表明氯諾昔康質量濃度在6.4~80 μg/mL范圍內與峰面積線性關系良好。

A.對照品;B.供試品;C.陰性對照。圖1 氯諾昔康干混懸劑的HPLC色譜圖Figure 1 HPLC chromatograms of lornoxicam dry suspension
2.6.7 精密度試驗 取對照品溶液(80 μg/mL),分別在同一日內連續進樣6次,及在6日內連續進樣6次,測定氯諾昔康的峰面積,測得日內精密度RSD值為1.26%,日間精密度RSD值為1.12%,表明儀器精密度良好。
2.6.8 加樣回收率試驗 精密稱取已知質量分數的同一批供試品(批號:2015061502)9份,每3份為一組,置于50 mL容量瓶中,分別加入相當于80%、100%、120%的氯諾昔康對照品溶液,加流動相定容,配制成高、中、低3個質量濃度各3份,搖勻,濾過,進樣,計算回收率。結果測得氯諾昔康的平均回收率為99.89%,RSD為1.02%。
2.6.9 樣品質量分數的測定 取不同批號的氯諾昔康干混懸劑按“2.6.3”項下方法制成供試品溶液,并按“2.6.1”項下色譜條件進樣測定。結果測得3批樣品的質量分數分別為99.2%、98.6%、98.3%,均大于95%,符合測定要求。
2.7 氯諾昔康干混懸劑溶出度的測定
采用漿法,分別選擇水、HCl(0.1 mol/L)、磷酸鹽緩沖液(pH=7.4)作為溶出介質,轉速設定在50 r/min,分別在5、10、15、20、30、45、60 min時間點取樣分析,同時加入同體積同溫度的溶出介質。氯諾昔康干混懸劑在以上3種介質中的溶出度曲線見圖2。可見,以水和HCl作為溶出介質時,氯諾昔康干混懸劑在20 min內均未完全溶出;在磷酸鹽緩沖液中溶出較快且平緩,且在25 min內能夠基本全部溶出。
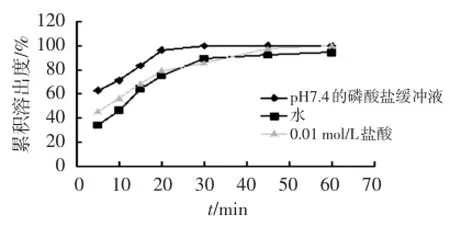
圖2 氯諾昔康干混懸劑溶出度曲線Figure 2 Dissolution curves of lornoxicam dry suspension
2.8 氯諾昔康干混懸劑穩定性研究
取3 批 樣 品 (2015061502、2015061506、2015061508)按照市售復合鋁箔袋包裝,于25℃、60%濕度條件下放置6個月,分別于0、1、2、3、6月末取樣檢測平均沉降體積比、再分散性、質量分數和20 min時的溶出度,結果見表5。可見,氯諾昔康干混懸劑在上述條件下放置6個月后,各項指標無明顯變化,穩定性較好。
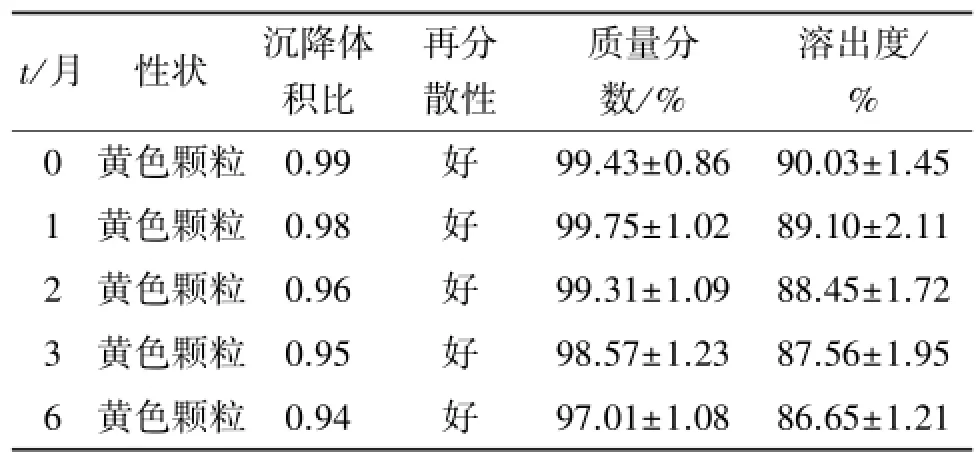
表5 3批樣品穩定性考察結果Table 5 The stability results of three batches of samples(n=3)
3 討論
干混懸劑是將藥物用適宜方法制成顆粒或粉末狀的制劑,使用時加水即迅速分散成混懸劑,增加了難溶性藥物在體內吸收和利用的速度和程度,提高了生物利用度[7]。干混懸劑的制備過程中選擇合適的助懸劑是關鍵的一步,達到合格的沉降體積比是制備干混懸劑的技術難點[8]。本文在選擇助懸劑時,對HPMC、黃原膠、CMC-Na進行考察,結果表明使用黃原膠和CMC-Na共同做為助懸劑時,所制得的混懸劑沒有沉淀,溶解性好,沉降體積比符合要求,再分散性好。
參照文獻[9]建立了測定氯諾昔康干混懸劑含量及溶出度的HPLC法,結果表明方法專屬性強、靈敏、快速,結果準確、重復性好。長期(6個月)穩定性試驗表明本文制備的氯諾昔康干混懸劑穩定性較好,更長時間的穩定性仍需進一步考察。
本研究結果為氯諾昔康干混懸劑的進一步研發提供了理論參考。
參考文獻:
[1]瞿浩,夏瑋.氯諾昔康的藥理及臨床應用[J].山東醫藥,2011,51(29):112-113.
[2]徐偉,陸軍.富馬酸氯馬斯丁干混懸劑的制備及穩定性研究[J].天津藥學,2004,16(2):27-29.
[3]周舍典,王莉,徐峰,等.55例患者應用氯諾昔康鎮痛效果及其不良反應回顧性調查[J].中國藥房,2005,16 (5):376-377.
[4]王忠云,黃亞輝,錢燕寧.氯諾昔康對胃癌患者術后曲馬多鎮痛的影響[J].臨床麻醉學雜志,2003,19(10):604-606.
[5]楊戒驕,向莉,李雙梅,等.頭孢克肟干混懸劑制備工藝及沉降體積比影響因素研究[J].中國藥房,2008,19(10):765-767.
[6]國家藥典委員會.中華人民共和國藥典:2015年版二部[M].北京:中國醫藥科技出版社,2015:附錄16.
[7]宋金春,陳杏.復方阿莫西林干混懸劑的制備與含量測定[J].中國藥師,2012,15(11):1606-1609.
[8]胡瑞標.頭孢克洛干混懸劑的研究[D].廣州:廣州中醫藥大學,2011.
[9]張建軍,高緣,樊偉明,等.高效液相色譜法測定氯諾昔康薄膜衣片的溶出度[J].中國醫院藥學雜志,2005,25 (5):447-448.
(責任編輯:陳翔)
中圖分類號:R944.3
文獻標志碼:A
文章編號:1006-8783(2016)03-0275-04
DOI:10.16809/j.cnki.1006-8783.2015120404
收稿日期:2015-12-04
基金項目:湖北省自然科學基金項目(2015CKB757)
作者簡介:沙崢(1989—),女,2014級碩士研究生,Email:384642983@qq.com;通信作者:張洪(1962—),男,教授,碩士生導師,從事消化系疾病治療藥物的藥劑學與藥理學研究,電話:027-88041911-88382,Email:zhanghongwhu@163.com。
Study on the preparation methodology and quality of lornoxicam dry suspension
SHA Zheng,ZHANG Hong,PENG Rui,ZHANG Ying,WEI Danyun
(Department of Pharmacy,Renmin Hospital of Wuhan University,Wuhan 430060,China)
AbstractObjective To study the preparation and quality of lornoxicam dry suspension.Methods The sedimentation rate and dispersion of lornoxicam dry suspension were investigated by single factor study.The prescription methodology was optimized by orthogonal design.HPLC method was used for determining the content of lornoxicam dry suspension.The dissolution behavior and stability of lornoxicam dry suspension were also been investigated.Results The optimal prescription methodology for lornoxicam dry suspension was as follows CMC-Na 8% and xanthan gun 5% as the suspending agents MCC 15% as the disintegrant and 15%PVP in 50%ethanol solution as the adhesive.The average labeled content of 3 batches of samples was 98.7%.The pH 7.4 phosphate buffer was the optimal dissolution media with fast dissolution rate and relatively smooth.The each index of suspension displayed no significant change by the stability test.Conclusion The optimized formulation process is simple feasible stable and repeatable.The preparation is in accordance with the quality requirements of dry suspension.
Key wordslornoxica dry suspension preparation methodology HPLC stability
