多重連接探針擴(kuò)增技術(shù)對(duì)1個(gè)假肥大型中國漢族肌營養(yǎng)不良癥家系的遺傳學(xué)研究*
2016-07-18 03:12:28楊佳良羅懷超朱世凱
國際檢驗(yàn)醫(yī)學(xué)雜志 2016年12期
關(guān)鍵詞:檢測
楊佳良,羅懷超,2,馬 誓,郝 芳,朱世凱,周 玉△
(1.電子科技大學(xué)附屬四川省人民醫(yī)院檢驗(yàn)科,成都 610072;2.瀘州醫(yī)學(xué)院臨床檢驗(yàn)系,四川瀘州 646000;3.電子科技大學(xué)附屬四川省人民醫(yī)院器官移植中心,成都 610072)
·論著·
多重連接探針擴(kuò)增技術(shù)對(duì)1個(gè)假肥大型中國漢族肌營養(yǎng)不良癥家系的遺傳學(xué)研究*
楊佳良1,羅懷超1,2,馬誓1,郝芳1,朱世凱3,周玉1△
(1.電子科技大學(xué)附屬四川省人民醫(yī)院檢驗(yàn)科,成都 610072;2.瀘州醫(yī)學(xué)院臨床檢驗(yàn)系,四川瀘州 646000;3.電子科技大學(xué)附屬四川省人民醫(yī)院器官移植中心,成都 610072)
目的擬通過多重連接探針擴(kuò)增技術(shù)(MLPA)對(duì)1個(gè)假肥大型進(jìn)行性肌營養(yǎng)不良(DMD)家系進(jìn)行檢測以明確DMD的診斷,結(jié)合患者臨床表現(xiàn)進(jìn)行分析并對(duì)MLPA技術(shù)用于DMD臨床診斷進(jìn)行探討。方法收集1個(gè)DMD患者家系,該家系共12人,男性患者2例。分離外周血并提取DNA,用MLPA的方法對(duì)其進(jìn)行基因診斷。結(jié)果先證者符合DMD診斷。基因檢測提示先證者和其弟弟攜帶抗肌萎縮蛋白基因缺失突變DMD(Exon3-11)。先證者母親為致病基因攜帶者。結(jié)論通過MLPA檢查技術(shù)明確了一個(gè)中國漢族DMD家系的致病基因,MLPA技術(shù)能夠用于DMD疾病的診斷。
假肥大型肌營養(yǎng)不良;家系;基因分析;多重連接探針擴(kuò)增技術(shù);抗肌萎縮蛋白基因
假肥大型進(jìn)行性肌營養(yǎng)不良(DMD)為X-連鎖隱性遺傳性疾病,男性多發(fā),女性多為致病基因的攜帶者。陽性患者的特點(diǎn)為進(jìn)行性對(duì)稱性肌無力,Gower征陽性,腓腸肌假性肥大。一般5歲前發(fā)病,6歲后出現(xiàn)行走困難,13歲喪失行走能力而依靠輪椅生活,20歲左右因心力衰竭而死亡。根據(jù)目前的研究,DMD發(fā)病多與遺傳因素有關(guān)。多重連接探針擴(kuò)增技術(shù)(MLPA)于2002年由Schouten等首先報(bào)道,是近幾年發(fā)展起來的一種針對(duì)DNA序列進(jìn)行定性和半定量分析的新技術(shù)。該技術(shù)高效、特異,在一次反應(yīng)中可以檢測45個(gè)核苷酸序列拷貝數(shù)的改變,目前已經(jīng)應(yīng)用于多個(gè)領(lǐng)域、多種疾病的研究。近年來報(bào)道DMD是位于X染色體短臂Xp21的抗肌萎縮蛋白基因(又稱dystrophin基因、DMD基因)突變所致。目前,在眾多確定與肌營養(yǎng)不良發(fā)生有關(guān)的致病基因中,抗肌萎縮蛋白基因備受關(guān)注。它是目前已知的人類最長的基因,包括79個(gè)外顯子。根據(jù)國外報(bào)道,其主要突變類型為基因部分缺失,占全部突變類型的60%~65%[1-3]。本研究對(duì)一個(gè)DMD家系采用多重連接基因探針擴(kuò)增技術(shù)進(jìn)行基因診斷和臨床研究,現(xiàn)報(bào)道如下。
1資料與方法
1.1一般資料2015年四川省人民醫(yī)院臨床初診為DMD的先證者和此家系其他成員,共3代12人。
1.2方法
1.2.1標(biāo)本采集在征得先證者及家屬知情同意后,采集5%乙二胺四乙酸(EDTA)抗凝靜脈血,-80 ℃保存,用試劑盒提取DNA,-20 ℃保存待用。選擇健康男性對(duì)照,健康女性對(duì)照,且對(duì)照均無心臟病、肌無力、肌萎縮和血清肌酶升高,無遺傳病家族史。家庭成員需簽署知情同意書。
1.2.2檢測方法(1)對(duì)所有研究對(duì)象進(jìn)行外周血DNA的提取。(2)MLPA的方法對(duì)先證者及家系成員進(jìn)行基因突變的研究和分析。MLPA即探針與靶序列DNA雜交,之后通過連接,PCR擴(kuò)增,產(chǎn)物通過毛細(xì)管電泳及數(shù)據(jù)收集進(jìn)行分析最后得出結(jié)論。通過每個(gè)探針的相對(duì)峰值比(RPR)來判斷標(biāo)本DMD目的片段的拷貝數(shù)。正常:0.7
1.3治療與隨訪對(duì)通過基因診斷確診的患者進(jìn)行適當(dāng)?shù)闹委熀碗S訪。
1.4統(tǒng)計(jì)學(xué)處理Coffalyser9.4軟件進(jìn)行數(shù)據(jù)處理及統(tǒng)計(jì)分析。
2結(jié)果
2.1患者及家系該家系來自四川省,家系圖見圖1,該家系中共有12人(不含先證者)。先證者的弟弟1993年2月出生,也出現(xiàn)雙下肢無力,雙側(cè)腓腸肌代償性肥大,于當(dāng)?shù)蒯t(yī)院診斷為DMD。另外先證者的妹妹2004年出生,未出現(xiàn)DMD的臨床表現(xiàn)。DMD的病因和發(fā)病機(jī)制復(fù)雜多變,結(jié)合該家系的特點(diǎn),可歸納如下:(1)多在男性身上發(fā)病,且多在5歲前發(fā)病;(2)首發(fā)癥狀通常會(huì)表現(xiàn)為行走慢、足尖著地、登梯困難、不能正常跑步,容易跌倒;肌無力自軀干和四肢近端開始緩慢進(jìn)展,下肢重于上肢;步態(tài)呈鴨步;攀登起立征(Gower征)陽性,為本病特征性表現(xiàn);(3)隨著病情的進(jìn)一步發(fā)展,近端肌肉有所萎縮,腓腸肌代償性肥大;(4)晚期患者的四肢、軀干、肩部肌肉均見明顯萎縮,最后因呼吸肌萎縮而出現(xiàn)呼吸困難、咳嗽無力,心律失常等,多在25~30歲以前死于呼吸道感染、心力衰竭或消耗性疾病。(5)輔助檢查:實(shí)驗(yàn)室檢查較特征性的改變?yōu)轱@著升高的血清肌酸激酶(CK)水平,患兒出生后其CK水平即可顯著升高,隨年齡的增長而逐漸上升,最高可達(dá)正常上限的100倍以上,到疾病中后期,因大部分肌纖維已被破壞,CK水平則逐漸下降。
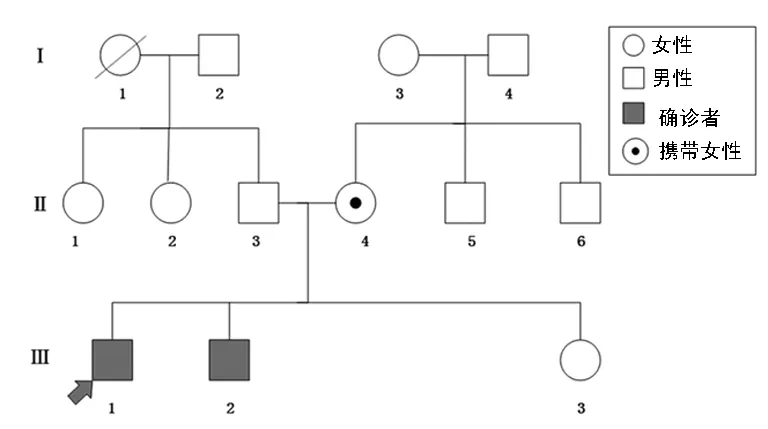
注:箭頭所示為先證者。
圖1家系圖
2.2臨床資料先證者,男,24歲,因“呼吸困難,雙下肢乏力”入院,血壓135/64 mm Hg,脈搏121次/分,呼吸18次/分,平車推入病房,呼叫睜眼,不發(fā)音,四肢遲緩。頭顱五官無畸形,全身皮膚黏膜無黃染,淺表淋巴結(jié)無腫大,鞏膜無黃染,雙側(cè)瞳孔等大等圓,對(duì)光反射靈敏。頸軟,無抵抗,頸靜脈無充盈怒張,已行氣管切開術(shù),人工氣道通暢,甲狀腺未捫及腫大,胸廓漏斗狀,聽診雙肺呼吸音清晰,未聞及濕啰音及哮鳴音。心界不大,心率121次/分,律齊,各瓣膜區(qū)聽診未聞及病理性雜音,全腹平軟,無壓痛,無肌緊張及反跳痛,肝脾肋下未觸及,未觸及包塊,肝頸靜脈回流征(-),移動(dòng)性濁音(-),膀胱叩診(-),腸鳴音無增強(qiáng)及減弱。已行導(dǎo)尿術(shù),尿管在位、通暢,雙下肢無水腫。四肢肌力0級(jí),生理反射及病理反射均未引出。先證者經(jīng)心肺復(fù)蘇后,因缺血缺氧性腦病于2015年5月25日死亡,死亡年齡24歲。
2.3DNA序列分析先證者Ⅲ-1和家系成員Ⅲ-2、Ⅱ-4均攜帶抗肌萎縮蛋白基因缺失突變DMD(Exon3-11),見圖2(見《國際檢驗(yàn)醫(yī)學(xué)雜志》網(wǎng)站主頁“論文附件”)。從圖中可以看出攜帶該基因缺失突變的男性外顯子DMD(3-11)全部缺失,其母親因?yàn)橛袃蓷lX染色體而表現(xiàn)出約50%的缺失。每個(gè)探針的RPR見表3,所有患者在外顯子3-11的RPR都為0,表明這些區(qū)域完全缺失并表現(xiàn)出DMD的典型臨床癥狀。其母親為攜帶者,RPR在0.5左右,并不表現(xiàn)出DMD的典型臨床表現(xiàn)。其他成員及正常男性和女性RPR在0.7~1.3為正常,未檢出攜帶此突變。
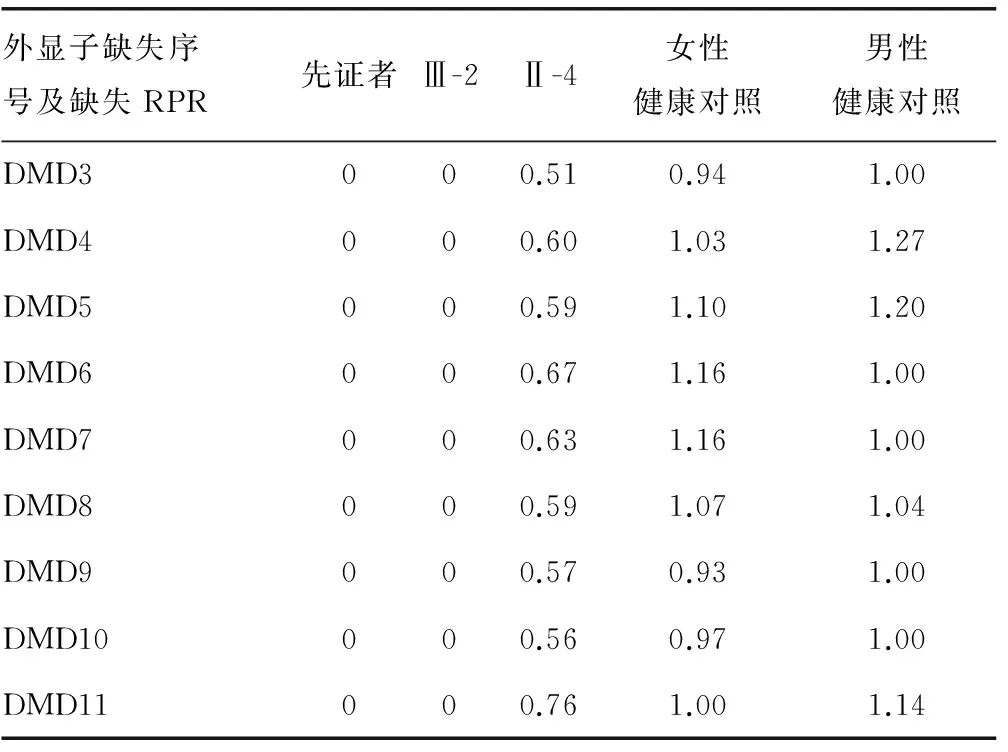
表3 MLPA探針的RPR
注:MLPA顯示家系中先證者Ⅲ-1,以及家系成員Ⅲ-2、Ⅱ-4均攜帶抗肌萎縮蛋白基因缺失突變DMD外顯子(3~11),其他家系成員及健康男女對(duì)照均未發(fā)現(xiàn)此突變。
3討論
DMD是最常見的一類X連鎖隱性遺傳病,以進(jìn)行性肌營養(yǎng)不良癥為主要臨床表現(xiàn)。主要是男性發(fā)病,女性為致病基因的攜帶者。本病的發(fā)生是由于編碼抗肌萎縮蛋白基因的突變所引起,該基因位于X染色體p21上,是人體最大的基因,長達(dá)2.3 Mb,含79個(gè)外顯子,79 個(gè)內(nèi)含子,編碼約14×103的RNA,翻譯產(chǎn)物為抗肌萎縮蛋白,它于肌細(xì)胞膜脂質(zhì)中,對(duì)穩(wěn)定細(xì)胞膜、防止細(xì)胞壞死自溶起重要作用。定量分析表示,DMD患者肌細(xì)胞內(nèi)抗肌萎縮蛋白近乎完全缺失,故該病通常5歲左右發(fā)病,肌進(jìn)行性萎縮,預(yù)后差。沒有切實(shí)有效的臨床治療方法。有約1/3病例為散發(fā),沒有家族史,是由基因新突變?cè)斐伞?/p>
本研究通過MLPA的方法對(duì)1例DMD家系進(jìn)行抗肌萎縮蛋白基因的全外顯子檢測,發(fā)現(xiàn)其母為DMD外顯子3~11缺失突變攜帶者,其所生兩個(gè)兒子也都為DMD外顯子(3~11)基因缺失,并有典型的DMD臨床表現(xiàn),在本醫(yī)院通過基因分析和一系列臨床檢查確診為DMD患者。其女并未檢出基因突變,也沒有DMD的典型臨床癥狀及實(shí)驗(yàn)檢查結(jié)果。
張曉[4]提出本病無明顯種族和地域的差異。但杜青[5]則通過單鏈構(gòu)象多態(tài)性分析(SSCP)篩查確診患者第48號(hào)外顯子存在明顯的異常,而提出中國人種DMD疾病的基因突變位點(diǎn)可能和外國人種存在著區(qū)別。而本研究通過MLPA的方法對(duì)DMD家系進(jìn)行抗肌萎縮蛋白基因的全外顯子研究,發(fā)現(xiàn)其母為DMD外顯子3~11缺失突變攜帶者,其所生兩個(gè)兒子也為DMD外顯子(3~11)基因缺失,并有典型的DMD臨床表現(xiàn),在本醫(yī)院通過基因分析和一系列臨床檢查確診為DMD患者,且先證者因心肺復(fù)蘇術(shù)后,缺血缺氧性腦病于2015年5月25日死亡,死亡年齡24歲。先證者妹妹并未檢出基因突變,也沒有DMD的典型臨床及實(shí)驗(yàn)檢查的表現(xiàn),也沒有發(fā)現(xiàn)48號(hào)外顯子的突變。正如目前已知的抗肌萎縮蛋白基因突變類型中最為常見的為缺失,存在2個(gè)缺失熱區(qū)。一個(gè)位于中央?yún)^(qū)域,多包括45~55號(hào)外顯子,另一個(gè)缺失熱區(qū)位于基因的5′端,多包括2~20號(hào)外顯子[6-7]。而本研究顯示3~11號(hào)外顯子丟失,符合上訴者的5′端的缺失熱點(diǎn)區(qū)。上述兩個(gè)缺失熱區(qū)中的外顯子缺失約占全部缺失突變的98%。有研究顯示于1~20號(hào)外顯子,21~40號(hào)外顯子,41~60號(hào)外顯子,61~79號(hào)外顯子上的點(diǎn)突變率分別為30.4%、30.4%、24.6%及14.5%[8]。所以,DMD發(fā)病的表現(xiàn)形式及基因突變的位置因人而異,在檢測DMD時(shí)只檢測熱點(diǎn)區(qū)域,或只查其中的一種突變是不夠的。
目前檢測DMD的方法主要有Southern印跡雜交、DMD基因多重PCR、短串聯(lián)重復(fù)序列PCR及反轉(zhuǎn)錄PCR。Chamberlain等[9]設(shè)計(jì)了9對(duì)引物(針對(duì)外顯子4、8、12、17、19、44、45、48、51)的PCR;Beggs等[10]增設(shè)了另外9對(duì)引物(針對(duì)外顯子3、6、13、43、47、50、52、60、49),這18對(duì)引物可檢出大部分的DMD基因缺失患者。但這種方法不能檢測DMD基因缺失型和重復(fù)型雜合子攜帶者。而MLPA技術(shù)可以全面地檢測抗肌萎縮蛋白基因79個(gè)外顯子的全部缺失和重復(fù)突變。該方法靈敏、特異,廣泛用于DMD患者和攜帶者的檢測[11-13]。但MLPA也有其自身的缺點(diǎn),即鄰近探針連接位點(diǎn)的微小突變或多態(tài)會(huì)影響探針雜交及連接,導(dǎo)致擴(kuò)增失敗;MLPA也無法檢測出X染色體的平衡易位及抗肌萎縮蛋白基因內(nèi)的微小突變[14]。因此對(duì)于單個(gè)外顯子缺失的情況,要用PCR和測序?qū)ζ溥M(jìn)行驗(yàn)證。但對(duì)于DMD的診斷還應(yīng)結(jié)合臨床表現(xiàn)、病理肌電、實(shí)驗(yàn)室檢查等綜合地來診斷該病。由于該病尚無更好的治療方法,所以切實(shí)有效的產(chǎn)前診斷和遺傳咨詢顯得尤為必要。
該家系中其母親為致病基因的攜帶者,而其所生兩個(gè)兒子均患DMD,且臨床表現(xiàn)非常典型:進(jìn)行性對(duì)稱性肌無力和腓腸肌代償性腫大。肌電圖、肌肉病理及實(shí)驗(yàn)室的檢查都符合DMD的診斷,與MLPA所得結(jié)果是一致的。且先證者的妹妹用MLPA的方法沒有檢測到基因突變,其也沒有DMD的臨床表現(xiàn),這更好地說明了MLPA是一種可以用于臨床DMD診斷和產(chǎn)前篩查的簡單、靈敏、特異的方法。正如上面所說MLPA也有其自身局限性。所以期待更多的家系報(bào)道,基因位點(diǎn)和突變類型的報(bào)導(dǎo)及基因相關(guān)功能的研究,更多的診斷及治療方法被發(fā)現(xiàn),為DMD的預(yù)防、診斷和治療開拓新篇章。
[1]Muntoni F,Torelli S,Ferlini A.Dystrophin and mutations:one gene,several proteins,multiple phenotypes[J].Lancet Neurol,2003,2(12):731-740.
[2]Prior TW,Bridgeman SJ.Experience and strategy for the molecular testing of Duchenne muscular dystrophy[J].J Mol Diagn,2005,7(3):317-326.
[3]Hu XY,Ray PN,Murphy EG,et al.Duplicational mutation at the Duchenne muscular dystrophy locus:its frequency,distribution,origin,and henotypegenotype correlation[J].Am J Hum Genet,1990,46(4):682-695.
[4]張曉.假肥大型肌營養(yǎng)不良癥7例臨床特征分析[J].中國社區(qū)醫(yī)師(醫(yī)學(xué)專業(yè)),2011,13(278):133.
[5]杜青.一個(gè)中國家系假肥大型肌營養(yǎng)不良相關(guān)基因dystrophin的突變位點(diǎn)的檢測[D].武漢:華中科技大學(xué),2013.
[6]Lai PS,Takeshima Y,Adachi K,et al.Comparative study on deletions of the dystrophin gene in three Asian populations[J].J Hum Genet,2002,47(10):552-555.
[7]Danieli GA,Mioni F,Müller CR,et al.Patterns of deletions of the dystrophin gene in different European populations[J].Hum Genet,1993,91(4):342-346.
[8]Takeshima Y,Yagi M,Okizuka Y,et al.Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center[J].J Hum Genet,2010,55(6):379-388.
[9]Chamberlain JS,Gibbs RA,Ranier JE,et al.Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification[J].Nucleic Acids Res,1988,16(1):11141-11156.
[10]Beggs AH,Koenig M,Boyce FM,et al.Detection of 98%of DMD/BMD gene deletions by polymerase chain reaction[J].Hum Genet,1990,86(1):45-48.
[11]Gatta V,Scarciolla O,Gaspari AR,et al.Identification of deletions and duplications of the DMD gene in affected males and carrier females by multiple ligation probe amplification(MI,PA)[J].Hum Genet,2005,117(1):92-98.
[12]Li H,Ding J,Wang W,et al.Combining approach with muhiplex PCR and MLPA to detect deletion and duplication in DMD patients,carriers,and prenatal diagnosis[J].Chin J Med Genet,2009,26(3):318-322.
[13]Marzese DM.Mampel A.Gomez IC,et al.Detection of deletions and duplications in the Duchenne muscular dystrophygene by the molecular method MLPA in the first Argentine affected families[J].Genet Mol Res,2008,84(3):223-233.
[14]Kozlowski P,Jasinjiyiska AJ,Kwiatkowski DJ.New applications and developments in the use of multiplex ligation-dependentprobe amplification[J].E1ectrophoresis,2008,29(23):4627-4636.
Genetic study of Duchenne Muscular Dystrophy in a Chinese family by Multiplex ligationdependent probe amplification technique*
YANGJialiang1,LUOHuaichao1,2,MAShi1,HAOFang1,ZHUShikai3,ZHOUYu1△
(1.DepartmentofClinicalLaboratory,SichuanProvincialPeople′sHospitalAffiliatedtoUniversityofElectronicScienceandTechnology,Chengdu,Sichuan610072,China;2.InspectionDepartment,LuzhouMedicalCollege,Luzhou,Sichuan646000,China;3.OrganTransplantCenter,SichuanProvincialPeople′sHospitalAffiliatedtoUniversityofElectronicScienceandTechnology,Chengdu,Sichuan610072,China)
ObjectiveTo diagnosis for Duchenne Muscular Dystrophy(DMD)in a pedigree by multiplex ligation dependent probe amplification(MLPA),and to analyze on the basis of clinical manifestations,to explore the application value of MLPA technology in diagnosis for DMD.MethodsA pedigree of DMD patients were recruited in this study,12 person in this pedigree including 2 DMD patients.All the person in this pedigree were isolated peripheral blood and extracted DNA,conducted genetic diagnosis using MLPA.ResultsThe propositus was confirmed as a DMD patients.Genetic testing prompted the propositus and his brother carrying dystrophin gene deletion mutation DMD (Exon3-11).Propositus′s mother was pathogenic gene carrier.ConclusionThe disease gene is confirmed in in a pedigree with DMD by MLPA,and MLPA technique could be used to diagnose DMD disease.
Duchenne Muscular Dystrophy;pedigree;gene analysis;multiplex ligation dependent probe amplification;dystrophin gene

國家自然科學(xué)基金資助項(xiàng)目(81400437)。
楊家良,女,碩士研究生,主要從事遺傳學(xué)研究。△
,Email:zhouyu422@yahoo.com。
10.3969/j.issn.1673-4130.2016.12.012
A
1673-4130(2016)12-1624-03
2016-01-11修回日期:2016-03-18)
猜你喜歡
中國設(shè)備工程(2022年12期)2022-07-11 04:33:00
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:36
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:34
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:50
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:48
