6個不同海域文蛤地理群體的親緣關系分析
2016-03-04 07:31:58陳愛華吳楊平姚國興蔡永祥
海洋漁業 2016年3期
關鍵詞:差異
王 超,陳愛華,曹 奕,吳楊平,張 雨,姚國興,蔡永祥
(1.上海海洋大學水產與生命學院,上海 201306;2.江蘇省海洋水產研究所,南通 226007)
6個不同海域文蛤地理群體的親緣關系分析
王 超1,2,陳愛華2,曹 奕2,吳楊平2,張 雨2,姚國興2,蔡永祥2
(1.上海海洋大學水產與生命學院,上海 201306;2.江蘇省海洋水產研究所,南通 226007)
為探究中國遼東半島海域(遼寧群體)、長江口及其兩翼海域(江蘇群體)、臺灣海峽西部海域(福建群體)、珠江口及其兩翼海域(廣東群體)、北部灣海域(廣西群體)以及日本伊勢灣海域(三重群體)6個不同海域文蛤(Meretrix meretrix)地理群體的親緣關系,采用18S rRNA基因、線粒體細胞色素氧化酶c亞基Ⅰ(COⅠ)及16S rRNA基因3種分子標記進行序列測定與分析。測序結果顯示,3種基因序列長度分別在1 854、658、596 bp左右;18S rRNA基因堿基組成無偏異,序列較為保守;COⅠ與16S rRNA基因A+T平均含量明顯大于G+C含量,符合線粒體基因組成特征。通過MegAlign軟件比對6個地理群體文蛤,序列相似百分比分別為99.7%~100.0%(18S)、91.7%~99.8%(COⅠ)、90.2%~99.8%(16S),其中三重文蛤序列差異最大;以文蛤屬的簾文蛤(Meretrix lyrata)作外群,采用MEGA 5.03軟件中相鄰連接法(NJ)構建系統發育樹顯示,我國沿海5個文蛤群體聚為一枝且與日本三重文蛤分開,自展值分別為67%(18S)、99%(COⅠ)、98%(16S);我國沿海5個文蛤群體中,遼寧、江蘇、福建文蛤群體首先聚在一起,其次是廣西文蛤群體,最后是廣東文蛤群體。研究結果表明,遼寧、江蘇、福建文蛤群體同源性較高,親緣關系最近;廣西文蛤群體、廣東文蛤群體與我國其它文蛤群體間遺傳差異較大,地理遺傳分化明顯;而日本三重文蛤與我國沿海文蛤可定為文蛤的2個地理亞種。
文蛤;18S rRNA基因;COⅠ基因;16S rRNA基因;系統發育
文蛤(Meretrix meretrix Linnaeus)隸屬于軟體動物門(Mollusca),瓣鰓綱(Lamellibranchia),簾蛤目(Veneroida),簾蛤科(Veneridae),文蛤屬,為廣溫、廣鹽性灘涂埋棲型雙殼貝類,廣泛分布于中國、日本、朝鮮、印度等國家。文蛤肉質鮮美,營養豐富,其生物活性成分具有抗腫瘤、抗氧化、降血脂等功效[1],具有較高的經濟價值。目前我國文蛤養殖以野生苗種或近親繁育苗種為主,隨著累代養殖,近交衰退、種質下降等現象明顯,導致養殖文蛤生長速度減慢、抗逆力下降。加之,不同海域天然苗種異地移養,促使不同遺傳背景文蛤群體基因交流增強,對文蛤增養殖業的健康發展十分不利。因此,文蛤種質資源研究與良種選育工作亟需開展,而從分子水平了解不同海域文蛤的遺傳背景,可為以上工作的開展提供科學的理論基礎。
目前關于我國文蛤群體間遺傳多樣性的研究,主要集中在局部地區或少數群體之間[2-5]。而我國海岸線漫長且地形復雜,文蛤在南北沿海均有分布,要想全面了解文蛤種質資源狀況,必須擴大研究范圍,搜集足夠多的文蛤群體。馮建彬等[6]曾采用RAPD技術對我國遼寧、山東、江蘇、福建、廣東、廣西文蛤群體進行遺傳分析,為文蛤的種質資源利用提供了參考。但近十年來,我國文蛤增養殖業迅速擴張,異地移養、人工育種等可能對不同地區文蛤的遺傳背景產生影響,對當前我國文蛤種質資源狀況需重新認識。日本文蛤具有色澤艷麗、生長速度快等優良性狀,且與我國文蛤有較高的雜交配合力[7],有望成為我國文蛤良種選育的優勢材料之一。因此,本文以我國遼寧丹東、江蘇南通、福建長樂、廣東珠海、廣西北海和日本三重6個地理群體文蛤為研究對象,采用細胞核18S rRNA基因及線粒體COⅠ、16S rRNA基因3種分子標記,希望能夠較為全面地了解不同海域文蛤的種質資源狀況,通過比較日本三重文蛤與我國沿海文蛤的遺傳差異,為我國文蛤良種選育提供理論基礎。
1 材料與方法
1.1 樣品采集
實驗用文蛤為2014年10月采自江蘇省文蛤良種場保存的不同地理群體文蛤,采集信息見表1。6個地理群體文蛤分別來自于中國遼東半島海域遼東丹東(遼寧群體)、長江口及其兩翼海域江蘇南通(江蘇群體)、臺灣海峽西部海域福建長樂(福建群體)、珠江口及其兩翼海域廣東珠海(廣東群體)、北部灣海域廣西北海(廣西群體)及日本伊勢灣海域三重(三重群體),具體采集地見圖1。各群體均取20 ind個體,殼長約4 cm。采活體至實驗室解剖,取閉殼肌組織,保存于95%的酒精中。
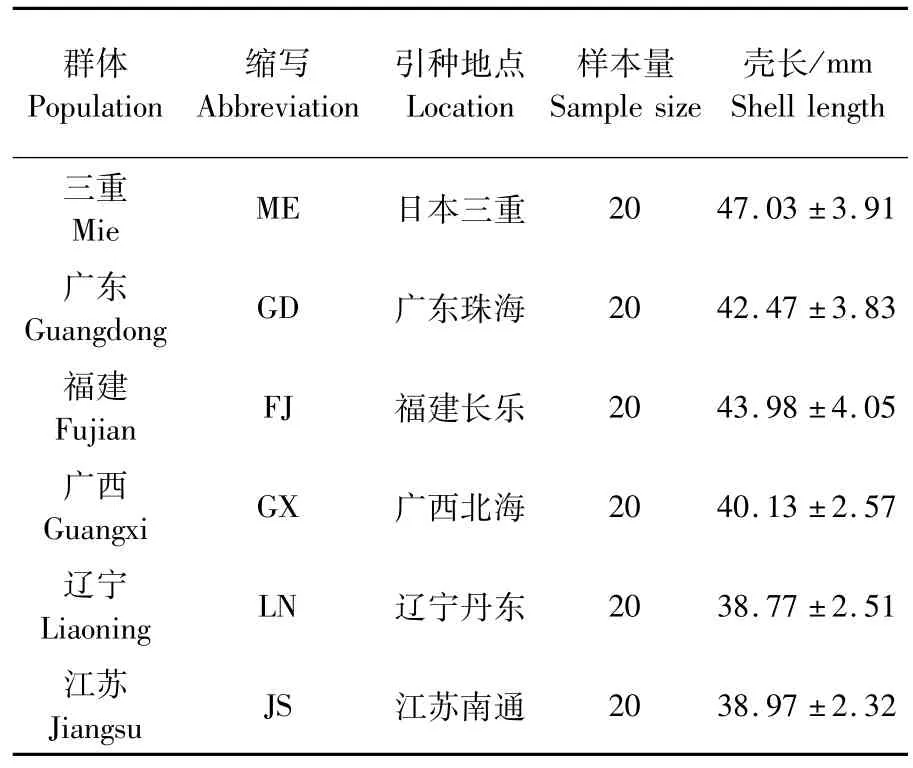
表1 不同地理群體文蛤樣品采集信息Tab.1 Information of different populations of M.meretrix
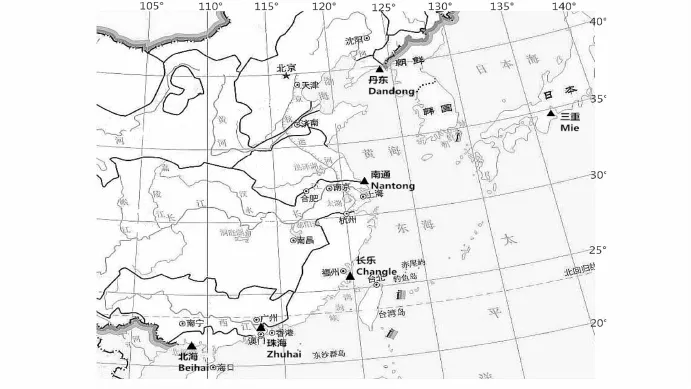
圖1 樣本采集地理分布圖(▲代表采樣地點)Fig.1 Geographic distribution of sam pling sites(▲shows a samp ling site)
1.2 DNA提取
取閉殼肌組織,采用傳統的酚/氯仿法提取總DNA,-20℃保存備用。
1.3 PCR擴增與產物測序
18S rRNA基因參考程漢良等[8]的方法設計3對引物分別擴增18S rRNA基因全序列的3個基因片段,再拼接為全序列;COⅠ基因采用無脊椎動物通用引物;16S rRNA基因引物參考ANDERSON[9]的序列。引物序列見表2,由生工生物工程(上海)股份有限公司合成。
PCR反應在Eppendorf PCR儀上進行,體系設置25μL。體系組成:模板DNA(50 ng· μL-1)1μL;上下游引物各1μL;10×PCR buffer 2.5μL;dNTP(2.5 mM)2μL;MgCl2(25 mM)2 μL;Taq DNA聚合酶(5 U·μL-1)0.4μL;ddH2O補足25μL。PCR反應條件設定:94℃預變性4 min;94℃變性40 s,58℃(18S)、52℃(COⅠ)、46℃(16S)退火30 s,72℃延伸1 min,35個循環;最后72℃延伸7 min。PCR產物經1%瓊脂糖凝膠電泳,EB染色,UV照射下割膠,Sanprep柱式DNA膠回收試劑盒回收純化。挑選效果理想的PCR純化產物送鉑尚生物技術(上海)有限公司使用擴增引物進行雙向測序。
1.4 序列分析
正反向序列用DNAStar Package(version 7.1.0)中的Seqman軟件組裝,結合測序峰圖進行人工校正。18S rRNA基因3個基因片段根據其重疊部分拼接為全序列。最終每個群體均得到18S rRNA、COⅠ、16S rRNA基因3條序列,用Editseq軟件編輯和分析,并除去兩端的引物部分。通過NCBI的Blast比對,驗證所得序列均為目的基因。MegAlign軟件比對序列,計算序列相似百分比與差異百分比。MEGA(version 5.03)分析堿基組成,計算變異位點數、簡約信息位點數,轉換(si)、顛換(sv)位點數及其比率(si/sv),并根據Kimura雙參數模型計算遺傳距離。以文蛤屬(Meretrix)的簾文蛤(Meretrix lyrata)相應序列作外群,GenBank序列號:JN996715.1(18S),JN898944.1(COⅠ),JN969948.1(16S),采用NJ法構建分子系統發育樹。
2 結果與分析
2.1 序列比對
對6個群體文蛤基因序列分析顯示,18S rRNA基因堿基平均含量分別是T 24.0%、C 24.2%、A 23.7%、G 28.1%、A+T 47.7%(表3)。日本三重文蛤的序列長度為1 849 bp,而我國沿海5個地理群體文蛤的序列長度均為1 854 bp,兩者相差5 bp是因為在位點231~235處存在5個堿基的插入/缺失(圖2)。由于18S rRNA基因具有較高的保守性,三重文蛤與我國文蛤僅有1個位點發生顛換(G/T),我國文蛤各群體間無變異發生。
COⅠ基因堿基平均含量為T 45.4%、C 13.6%、A 20.6%、G 20.4%、A+T 66.0%(表3)。序列不存在堿基的插入/缺失,長度均為658 bp(圖3),共有73個變異位點,其中簡約信息位點30個,平均轉換位點29個,平均顛換位點2個,比值R=si/sv=13.12。變異位點大多發生于密碼子的第三位,由于密碼子的簡并性,COⅠ基因編碼的219個氨基酸中只有3個位點發生變異。
16S rRNA基因堿基平均含量T 37.0%、C 12.0%、A 28.9%、G 22.2%、A+T 65.9%(表3)。序列共出現15個堿基的插入/缺失,分布于250~360之間的7個位點,三重文蛤、廣東文蛤、福建文蛤、廣西文蛤、遼寧文蛤、江蘇文蛤的序列長度依次為597、593、595、596、596、596 bp(圖4),共有變異位點71個,簡約信息位點11個,平均轉換位點20個,平均顛換位點7個,比值R=si/sv=2.94。

表2 引物序列Tab.2 Primer sequences
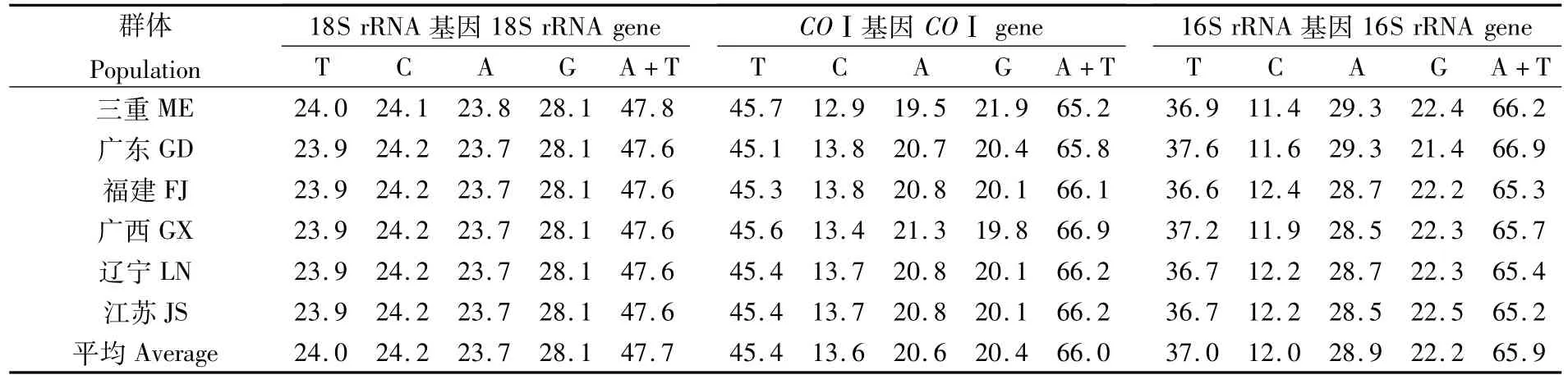
表3 不同地理群體文蛤18S rRNA、16S rRNA和COⅠ基因片段堿基組成百分比Tab.3 The base com position(%)of 18S rRNA,16S rRNA and COⅠgene fragments in different populations of M.meretrix (%)

圖2 不同地理群體文蛤18S rRNA基因序列比對Fig.2 Sequence alignment of partial 18S rRNA gene fragment in different populations of M.meretrix
用MegAlign軟件采用Jotun Hein方法對6個文蛤群體的18S rRNA基因、COⅠ基因及16S rRNA基因序列分別進行比對,序列相似百分比與差異百分比見表4。18S rRNA基因序列三重文蛤與我國沿海文蛤差異百分比為0.3%,我國各群體間相似百分比均達100.0%。COⅠ基因序列6個文蛤群體相似百分比91.7%~99.8%,三重文蛤與我國文蛤差異均達8.0%及以上,我國遼寧、江蘇、福建文蛤群體之間序列相似度較高,與廣西、廣東文蛤群體差異相對較大。16S rRNA基因序列6個群體相似百分比90.2%~99.8%,三重文蛤與我國文蛤差異度水平均達10.0%以上,序列相似度也是遼寧、江蘇、福建文蛤群體較高,其次是廣西、廣東文蛤群體。3種基因序列分析結果較為一致,我國遼寧、江蘇、福建文蛤群體同源性較高,與廣西、廣東文蛤之間差異明顯,三重文蛤與我國文蛤間基因序列差異最大。
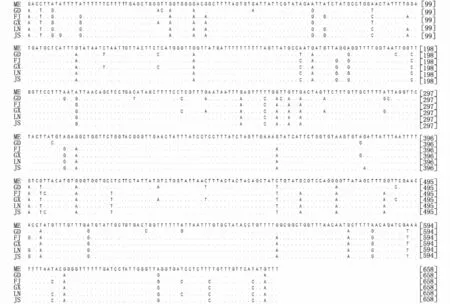
圖3 不同地理群體文蛤COⅠ基因序列比對Fig.3 Sequence alignment of partial COⅠgene fragment in different populations of M.meretrix

表4 基于Jotun Hein法計算6個地理群體文蛤18S rRNA基因、COⅠ基因、16S rRNA基因序列相似百分比與差異百分比Tab.4 Percentage of sequence identity and divergence of 18S rRNA,16S rRNA and COⅠgene in different populations of M.meretrix by Jotun Hein method (%)
用MegAlign軟件采用Jotun Hein方法對6個文蛤群體的18S rRNA基因、COⅠ基因及16S rRNA基因序列分別進行比對,序列相似百分比與差異百分比見表4。18S rRNA基因序列三重文蛤與我國沿海文蛤差異百分比為0.3%,我國各群體間相似百分比均達100.0%。COⅠ基因序列6個文蛤群體相似百分比91.7%~99.8%,三重文蛤與我國文蛤差異均達8.0%及以上,我國遼寧、江蘇、福建文蛤群體之間序列相似度較高,與廣西、廣東文蛤群體差異相對較大。16S rRNA基因序列6個群體相似百分比90.2%~99.8%,三重文蛤與我國文蛤差異度水平均達10.0%以上,序列相似度也是遼寧、江蘇、福建文蛤群體較高,其次是廣西、廣東文蛤群體。3種基因序列分析結果較為一致,我國遼寧、江蘇、福建文蛤群體同源性較高,與廣西、廣東文蛤群體之間差異明顯,三重文蛤與我國文蛤間基因序列差異最大。
2.2 遺傳距離與系統發育
用MEGA軟件根據Kimura 2-parametermodel計算包括外群在內的遺傳距離(D),用Bootstrap方法(1 000 replications)計算標準誤差。由18S rRNA基因計算得出的遺傳距離中(表5),三重文蛤與我國文蛤的遺傳距離D=0.001,與簾文蛤之間D=0.007;我國文蛤群體間遺傳距離為0,與簾文蛤之間D=0.008。
有研究表明,簾蛤科貝類DNA條形碼間隙區為0.040~0.100[10]。本研究根據COⅠ基因得出的遺傳距離中(表6),三重文蛤與我國江蘇(0.084)、廣西(0.091)、遼寧(0.080)、福建(0.082)、廣東(0.087)文蛤群體遺傳距離處于間隙區內且靠近0.100水平,已接近屬內種間差異;我國江蘇、廣西、福建、遼寧文蛤群體之間遺傳距離0.006~0.028,皆小于條形碼間隙0.040水平,即屬于種內差異;廣東文蛤群體與江蘇、遼寧、福建文蛤群體遺傳距離(0.066)處于間隙區且靠近0.040,與廣西文蛤群體距離0.036,可視為種內差異。
16S rRNA基因中(表7),三重文蛤與我國文蛤遺傳距離為0.092~0.098;我國遼寧、江蘇、福建、廣西文蛤群體遺傳距離較小(0.003~0.016),與廣東文蛤群體遺傳距離為0.039~0.054。THORP[11]提出群體間16S rRNA基因的遺傳距離是0.030~0.200,可見6個文蛤群體遺傳差異并沒有達到種間水平,遼寧、江蘇、福建文蛤群體甚至低于群體間水平。

圖4 不同地理群體文蛤16S rRNA基因序列比對Fig.4 Sequence alignment of partial 16S rRNA gene fragment in different populations of M.meretrix
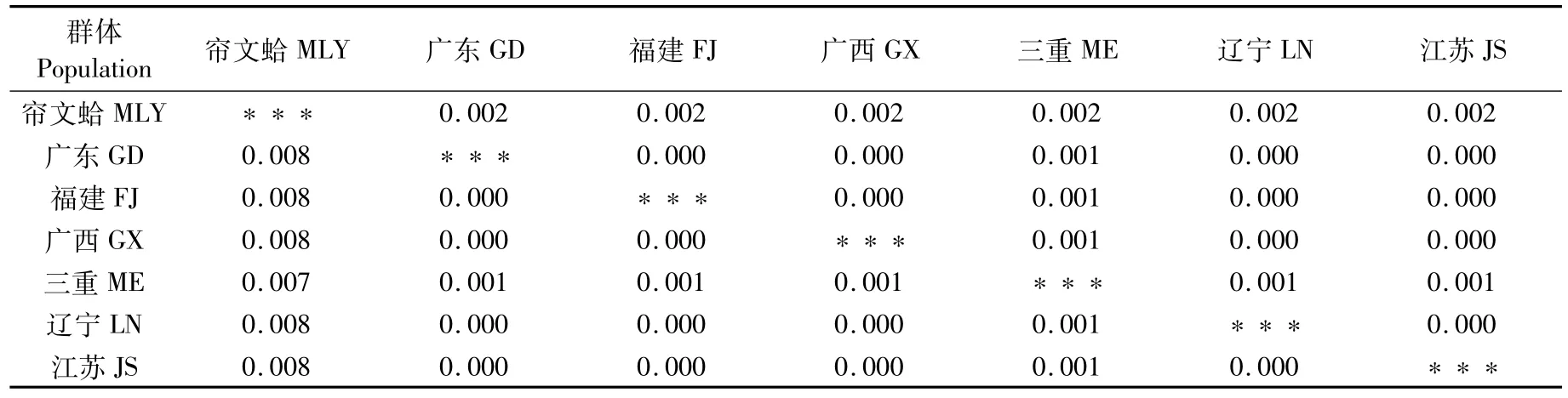
表5 6個地理群體文蛤及外群18S rRNA基因序列之間的遺傳距離和標準誤差Tab.5 Genetic distances(D)and corresponding standard errors of 18S rRNA gene among six populations of M.meretrix and outgroup
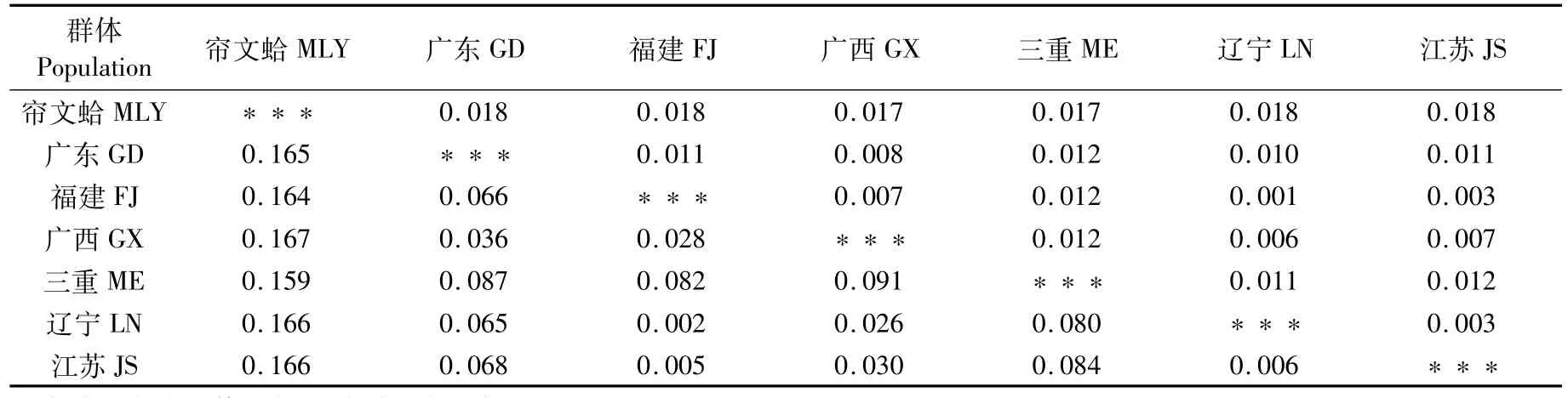
表6 6個地理群體文蛤及外群COⅠ基因序列之間的遺傳距離和標準誤差Tab.6 Genetic distances(D)and corresponding standard errors of COⅠgene among six populations of M.meretrix and outgroup
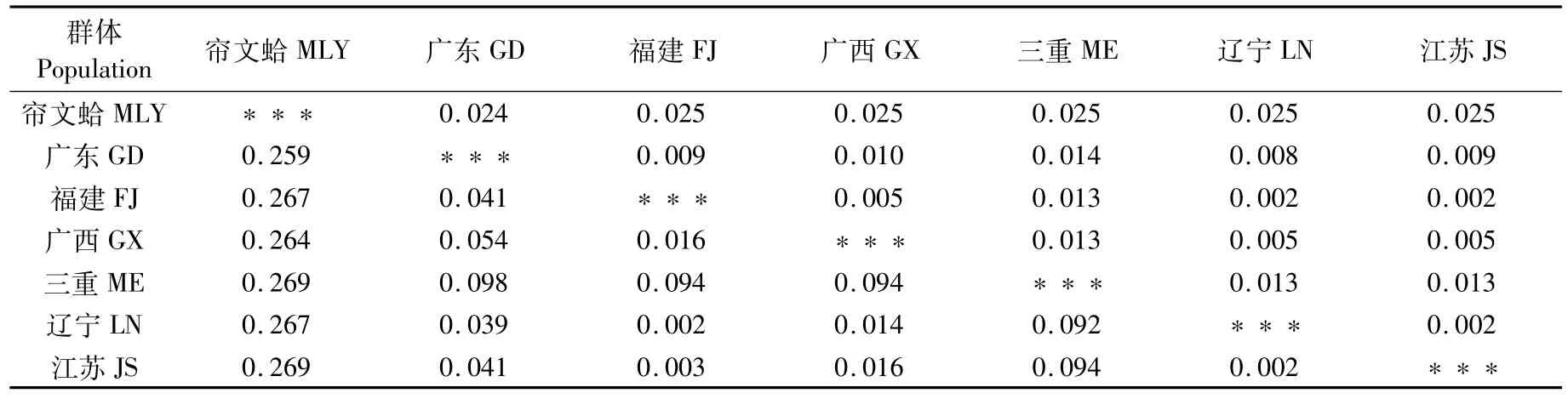
表7 6個地理群體文蛤及外群16S rRNA基因序列之間的遺傳距離和標準誤差Tab.7 Genetic distances(D)and corresponding standard errors of 16S rRNA gene am ong six populations of M.meretrix and outgroup
以簾文蛤作外群,采用相鄰連接法(NJ法)分別構建不同海域文蛤18S rRNA基因、COⅠ基因、16S rRNA基因序列的系統發育樹,節點數值表示Bootstrap 1 000次檢驗自展值。18S rRNA基因序列NJ樹拓撲結構顯示(圖5),6個地理群體文蛤首先聚為一枝與外群分開;文蛤種群中,我國沿海文蛤聚為一枝與三重文蛤分開,得到67%的支持率。COⅠ基因與16S rRNA基因序列的NJ樹拓撲結構一致(圖6-7),首先是6個文蛤種群聚為一枝與外群分開;其次我國沿海文蛤聚為一枝與三重文蛤分開,自展值分別高達99%(COⅠ)與98%(16S);我國不同地區文蛤中,遼寧、江蘇、福建文蛤群體親緣關系較近聚在一起,自展值分別是65%(COⅠ)與97%(16S),其次是廣西文蛤群體,最后是廣東文蛤群體。

圖5 基于18S rRNA基因序列的不同地理群體文蛤NJ樹(K imura 2-parameter model,1 000 replications)Fig.5 NJ phylogenetic tree based on 18S rRNA gene of different populations of M.meretrix
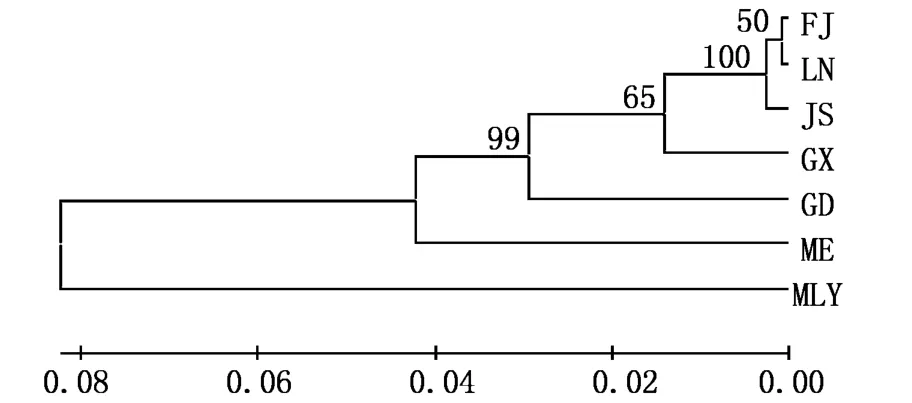
圖6 基于COⅠ基因序列的不同地理群體文蛤NJ樹(K imura 2-parameter model,1 000 replications)Fig.6 NJ phylogenetic tree based on COⅠgene of different populations of M.meretrix
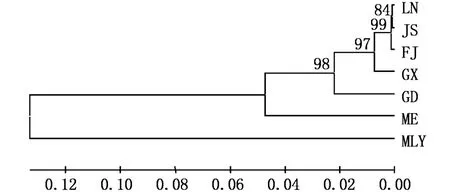
圖7 基于16S rRNA基因序列的不同地理群體文蛤NJ樹(K imura 2-parameter model,1 000 replications)Fig.7 NJ phylogenetic tree based on 16S rRNA gene of different populations of M.meretrix
2.3 不同文蛤群體的親緣關系
由以上分析可知,我國沿海遼寧、江蘇、福建文蛤群體遺傳分化低于地理群體間變異水平,親緣關系最近;廣西文蛤群體、廣東文蛤群體與我國其它文蛤群體間遺傳差異較大,地理遺傳分化明顯;日本三重文蛤與我國文蛤之間3種基因序列均有較大差異,按照簾蛤科貝類0.040~0.100條形碼間隙以及16S rRNA基因群體間0.030~0.200的遺傳距離,三重文蛤與我國文蛤之間超出種內分化但未達到種間水平,可認為二者達到地理亞種分化,3種基因序列拓撲結構均把我國文蛤群體聚為一枝與三重文蛤分開,且可信度較高,也支持把三重文蛤與我國文蛤定為文蛤的2個地理亞種。
3 討論
3.1 3種分子標記相結合應用于文蛤種群的親緣關系分析
18S rRNA基因在生物體內含量較大且在進化過程中具有保守性,適合不同層次系統發育的研究;COⅠ基因是線粒體氧化呼吸鏈的重要成員,基因變異較大;16S rRNA基因是非編碼蛋白質基因,不受密碼子編碼的選擇壓力影響,進化速度適中。本研究結合18S rRNA基因、COⅠ與16S rRNA基因分析不同地理群體文蛤的遺傳差異,利用3種基因序列不同的保守性,在聚類分析中得到了一致的拓撲結構,能夠有效反映不同地理群體文蛤的親緣關系。根據本文研究,18S rRNA基因堿基平均含量分別是T 24.0%、C 24.2%、A 23.7%、G 28.1%,4種堿基基本無偏異,與程漢良等[8]發表的簾蛤科貝類堿基組成一致;COⅠ與16S rRNA基因堿基A+T含量明顯高于G+C含量,符合線粒體基因組成特征。COⅠ基因共有73個變異位點,其中簡約信息位點30個,16S rRNA基因有71個變異位點,簡約信息位點11個,表明16S rRNA基因比COⅠ基因序列保守,與頭足綱(Cephalopoda)[12]、鮑科(Haliotidae)[13]等多個研究結果一致。
3.2 日本三重文蛤與我國沿海文蛤之間遺傳分化已達到亞種水平
DNA條形碼是HEBERT等[14]提出的一種基于COⅠ基因進行物種鑒定的分子技術。隨著研究的深入,HEBERT等[15]、BURNS等[16]提出以條形碼間隙(種內基因序列的最大遺傳距離與種間基因序列的最小遺傳距離之間的空隙)界定物種。陳軍[10]在簾蛤科貝類的DNA條形碼研究中,把0.040~0.100定義為簾蛤科的條形碼間隙,據此將315個樣品中的289個成功鑒定到種,并在BOLD數據庫中驗證了此間隙區的存在。因此,本研究以0.040~0.100的條形碼間隙探討不同群體文蛤的親緣關系是合理的。
在以往的研究中,DNA序列差異通常也被用來鑒定物種。有研究表明,馬蹄螺科(Trochidae)屬內種間COⅠ基因序列差異為5.7%~12.1%[17];珍珠蚌科(Margaritiferidae)屬內種間COⅠ基因序列差異11.57%~12.33%[18]。簾蛤科貝類的研究中,波紋巴非蛤(P.undulata)與織錦巴非蛤(P.textile)16S rRNA基因序列差異7.8%~8.0%,薄片鏡蛤(D.corrugate)與薄殼鏡文蛤(D.angulosa)序列差異為10.7%,兩者都被認為是相互獨立的種,防城港文蛤與其它地理群體文蛤COⅠ基因序列差異達到6.6%,被研究者認為具有兩個地理亞種的可能性[19]。本研究中日本三重文蛤與我國沿海文蛤COⅠ基因序列平均差異8.4%,16S rRNA基因序列平均差異10.4%,已接近種間分化水平。在聚類分析中,COⅠ基因與16S rRNA基因具有高度一致性,我國5個地理群體文蛤聚在一起,可信度達到99%(COⅠ)、98%(16S),三重文蛤則單獨形成一個分枝,即使在保守性較高的18S rRNA基因聚類中,三重文蛤也單獨形成一枝與我國沿海文蛤分開。
綜上,作者認為可將三重文蛤與我國沿海文蛤定為文蛤的2個地理亞種。吳楊平等[20]基于形態分析研究認為,日本三重文蛤與麗文蛤(Meretrix cusoria)相近,應為麗文蛤,并與福建長樂麗文蛤達到亞種水平。而潘寶平等[21]、CHEN等[22]、程漢良等[19]的研究則支持將麗文蛤訂為文蛤的同物異名或地理亞種的觀點。因此,本文將日本三重文蛤與我國沿海文蛤定為文蛤的2個地理亞種的結論與上述研究有一定的相似之處。
3.3 我國文蛤種質資源的現狀與保護
馮建彬等[6]的研究結果顯示,遼寧、山東、江蘇文蛤群體親緣關系較近,其次是福建、廣西、廣東群體。而在本研究中,福建文蛤群體與遼寧、江蘇文蛤群體聚在一起,其次是廣西、廣東群體,可見福建文蛤群體與遼寧、江蘇文蛤群體間親緣關系增近,我國其它文蛤群體間親緣關系并未發生較大改變。福建與遼寧、江蘇文蛤群體可能是由于近些年異地移養、苗種引入等造成了種質混雜,其它文蛤群體種質保護較好。日本三重縣(伊勢灣)與我國存在地理隔離,兩地氣候及海水理化因子差異較大,長期的自然選擇造成三重文蛤與我國文蛤達到亞種分化。為保證我國文蛤種質資源的可持續利用及增養殖業的健康發展,必須加強實施保護與改良措施,如:文蛤的增養殖中應避免近親繁殖,減少累代養殖;合理引種,文蛤異地移養要嚴格監管,避免對當地自然種群的基因污染;開展文蛤種質資源調查工作,對未受污染的種群建立種質資源保護區,對野生種已經大量減少的海域進行保種及人工增殖。
[1] 杜正彩,侯小濤,黃 慶,等.文蛤化學成分與藥理作用研究進展[J].安徽農業科學,2014,42(2):439-441.
DU Z C,HOU X T,HUANG Q,et al.Research progress of chemical composition and pharmacological activity ofMeretrix meretrix[J].Journal of Anhui Agricultural Sciences,2014,42(2):439-441.
[2] 杜曉東,鄧岳文,葉富良,等.廣東和廣西地區野生文蛤的遺傳多樣性[J].中國水產科學,2004,11(1):41-47.
DU X D,DENG Y W,YE F L,et al.Genetic diversity of seven wild populations ofMeretrix meretrix[J].Journal of Fishery Sciences of China,2004,11(1):41-47.
[3] 劉 馨,孫祥山,高悅勉.文蛤北方種群生化遺傳結構與變異的研究[J].水產科學,2006,25(4):179-183.
LIU X,SUN X S,GAO Y M.Genetic Structure and Variation inMeretrix meretrixfrom Northern China[J].Fisheries Science,2006,25(4):179-183.
[4] 林志華,董迎輝,李 寧,等.基于形態參數和AFLP標記的文蛤(Meretrix meretrix)不同地理群體遺傳變異分析[J].海洋與湖沼,2008,39(3):245-251.
LIN Z H,DONG Y H,LIN,et al.The genetic structure and diversity analysis of different geographical populations ofMeretrix meretrixusing morphological parameters and AFLP markers[J].Oceanologia Et Limnologia Sinica,2008,39(3):245-251.
[5] 董迎輝,姚定余,林志華,等.浙江和廣西兩種文蛤的分子鑒定及形態特征分析[J].水產學報,2011,35(10):1505-1502.
DONG Y H,YAO D Y,LIN Z H,et al.Molecular classification and morphological traits of two species ofMeretrix(Mollusca,Bivalvia)[J].Journal of Fisheries of China,2011,35(10):1505-1502.
[6] 馮建彬,李家樂,王美珍,等.我國沿海不同群體文蛤遺傳差異RAPD分析[J].海洋漁業,2005,27(4):281-285.
FENG J B,LI J L,WANG M Z,et al.RAPD analysis of genetic variations among the seven populations ofMeretrix meretrixin China’s coastal zone[J].Marine Fisheries,2005,27(4):281-285.
[7] 吳楊平,陳愛華,姚國興,等.3個不同地理群體紅殼色文蛤雜交的配合力分析[J].海洋漁業,2014,36(4):314-319.
WU Y P,CHEN A H,YAO G X,et al.Combining ability analysis on diallel cross from three different redMeretrix meretrixpopulations[J].Marine Fisheries,2014,36(4):314-319.
[8] 程漢良,彭永新,王 芳,等.6種簾蛤科貝類18S rRNA基因全序列比較分析[J].中國水產科學,2008,15(4):559-567.
CHENG H L,PENG Y X,WANG F,et al.Sequence analysis of 18S rRNA gene of six Veneridae clams(Mollusca:Bivalvia)[J].Journal of Fishery Sciences of China,2008,15(4):559-567.
[9] ANDERSON FE.Phylogeny and historical biogeography of the Loliginid squids(Mollusca:Cephyalopoda)based on mitochondrial DNA sequence data[J].Molecular Phylogenetics and Evolution,2000,15(2):191-214.
[10] 陳 軍.簾蛤科貝類分子系統學研究[D].青島:中國海洋大學博士學位論文,2012.
CHEN J.Molecular Systematics of Veneridae(Bivalvia,Mollusca)[D].Qingdao:Doctoral Dissertation of Ocean University of China,2012.
[11] THORP J P.The molecular dock hypothesis:Biochemical evaluation,genetic differentiation and systematics[J].Annual Review of Ecology and Systematics,1982,13(1):139-168.
[12] LIN X Z,ZHENG X D,XIAO S,et al.Phylogeny of the cuttlefishes(Mollusca:Cephalopoda)based on mitochondrialCOⅠand 16S rRNA gene sequence data[J].Acta Oceanologica Sinica,2004,23(4):699-707.
[13] AN H S,JEE Y J,MIN K S,et al.Phylogenetic analysis of six species of Pacific abalone(Haliotidae)based on DNA sequences of16S rRNA and cytochrome oxidase subunit I mitochondrial genes[J].Marine Biotechnology,2005,7(4):373 -380.
[14] HEBERT PD N,CYWINSKA A,BALL SL,etal.Biological identifications through DNA barcodes[J].Proceedings of the Royal Society of London B,2003(270):313-322.
[15] HEBERT PD N,RATNASINGHAM S,DEWAARD J R.Barcoding animallife:cytochrome c oxidase subunit I divergences among closely related species[J].Proceedings of the Royal Society of London B,2004(270):S96-S99.
[16] BURNS JM,JANZEN D H,HAJIBABAEIM,et al.DNA barcodes and cryptic species of skipper butterflies in the genus Perichares in Area de Conservacion Guanacaste,Costa Rica[J].Proceedings of the National Academy of Sciences of the United States of America,2008,105(17):6350-6355.
[17] DONALD K M,KENNEDY M,SPENCER H G.The phylogeny and taxonomy of australmonodontine topshells(Mollusca:Gastropoda:Trochidae),inferred from DNA sequences[J].Molecular Phylogenetics and Evolution,2005,37(2):474-483.
[18] MACHORDOM A,ARAUJO R,ERPENBECK D,et al.Phylogeography and conservation genetics of endangered European Margaritiferidae(Bivalvia:Unionoidea)[J].Biological Journal of the Linnean Society,2003,78(2):235-252.
[19] 程漢良,周旻純,陳冬勤,等.基于16S rDNA序列的簾蛤科貝類分子系統發育研究[J].水產科學,2012,31(11):657-662.
CHENG H L,ZHOU M C,CHEN D Q,et al.Phylogenetic Analysis of Veneridae(Mollusca:Bivalvia)based on Mitochondrial 16S rDNA[J].Fisheries Science,2012,31(11):657-662.
[20] 吳楊平,姚國興,陳愛華,等.文蛤屬2種貝類多變量形態分析及日本文蛤的物種有效性[J].水產學報,2011,35(9):1410-1417.
WU Y P,YAOG X,CHEN A H,etal.Multivariate morphometric analysis of two species ofMeretrix,withnotes on JapaneseMeretrix lusoria’s validity[J].Journal of Fisheries of China,2011,35(9):1410-1417.
[21] 潘寶平,吳琪,張素萍,等.文蛤屬(Meretrix)16S rRNA基因及ITS1序列的系統學分析[J].海洋與湖沼,2006,37(4):342-347.
PAN B P,WU Q,ZHANG S P,et al.Molecular phylogeny ofMeretrix(mollusca,bivalvia)based on 16s rRNA genes and ITS1 sequences[J].Oceanologia Et Limnologia Sinica,2006,37(4):342-347.
[22] CHEN A H,LI Z X,FENG G N.Phylogcnctic relationships of the genusMeretrix(Mollusca:Veneridae)based on mitochondrialCOⅠgene sequences[J].Zoological Research,2009,30(3):233-239.
Genetic relationship analysis of six Meretrix meretrix populations from different sea areas
WANG Chao1,2,CHEN Ai-hua2,CAO Yi2,WU Yang-ping2,ZHANG Yu2,YAO Guo-xing2,CAIYong-xiang2
(1.College of Fisheries and Life Science,Shanghai Ocean University,Shanghai201306;2.Jiangsu Marine Fisheries Research Institute,Nantong226007)
In this paper,18S ribosomal RNA gene,mitochondrial cytochrome oxidase c subunit I(COⅠ)gene and 16S rRNA gene sequences of six differentMeretrixmeretrixpopulations from Liaodong Peninsula sea area(Liaoning population),the Yangtze Estuary and its twowing sea areas(Jiangsu population),Western sea area of Taiwan Strait(Fujian population),Pearl River Estuary and its two wing sea areas(Guangdong population),Beibu Bay sea area(Guangxi population)and Ise Bay sea area,Japan(Mie population)were sequenced and analyzed to study their genetic relationship.The genetic background of differentMeretrix meretrixpopulations was analyzed in molecular level,to provide theoretical basis for germplasm resources research and breeding.Results of sequence analysis showed that:the length of three genetic sequences was about 1 854 bp,658 bp and 596 bp respectively;base composition of18S rRNA genewas unbiased,and its sequence was conservative;COⅠgene had 73 variable sites,among which 30 were parsim-informative sites,the average content of A+T was 66.0%;16S rRNA gene had 71 variable sites,among which 11 were parsim-informative sites,the average contentof A+Twas65.9%.A+T contentwas obviously higher than G+C content in bothCOⅠand 16S rRNA gene,which was in accordance with features of mitochondrial genes.Gene sequences of sixMeretrix meretrixpopulations were aligned by MegAlign,the percentage of sequence identity was 99.7%-100.0%(18S),91.7%-99.8%(COⅠ),90.2%-99.8%(16S)respectively.Meretrix meretrixfrom Mie had the largest difference.Phylogenetic trees were constructed by MEGA5.03 using adjacent connection method(NJ)with Meretrix lyrata as outgroup.Five Meretrix meretrix populations from China coast areas were clustered into one clade and separated fromMeretrix meretrixfrom Mie,Japan,bootstrapping value was 67%(18S),99%(COⅠ)and 98%(16S)respectively;among the five populations from China coast areas,Meretrix meretrixfrom Liaoning,Jiangsu,Fujian clustered together first,and then the population from Guangxi,the last was the population from Guangdong.Results indicated that:Meretrixmeretrixfrom Liaoning,Jiangsu,Fujian had a higher homology and their genetic relationship was the closest;a larger genetic difference was found amongMeretrixmeretrixfrom Guangxi,Guangdong and other populations from China,and their geographic genetic differentiation was obvious;Meretrixmeretrixfrom Mie,Japan and China coast areas could be described as two geographic subspecies.
Meretrixmeretrix;18S rRNA gene;COⅠgene;16S rRNA gene;phylogeny
Q 951
A
1004-2490(2016)03-0262-11
2015-09-12
江蘇省科技廳重點研發項目(BE2015324);江蘇省水產三新工程重大項目(D2014-16);江蘇省屬公益類科研院所能力提升項目(BM2015017);江蘇省水產良種保種和親本更新項目(BZ2014,2015);南通市農業科技創新項目(HL2014007)
王 超(1990-),男,河南信陽人,碩士研究生,主要從事貝類增養殖學研究。
E-mail:wangchao7198@163.com
陳愛華,研究員。Tel:0513-85228272,E-mail:chenah540540@aliyun.com
猜你喜歡
英語世界(2023年10期)2023-11-17 09:19:16
汽車實用技術(2022年10期)2022-06-09 11:16:58
音樂探索(2022年2期)2022-05-30 21:01:37
收藏界(2019年3期)2019-10-10 03:16:40
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
中國特種設備安全(2018年11期)2019-01-08 02:08:32
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
中國非營利評論(2017年1期)2017-11-09 03:09:10
海外華文教育(2017年8期)2017-11-07 04:42:02
現代語文(2016年21期)2016-05-25 13:13:50
