(Z)-2-(2-三苯甲基氨基噻唑-4-基)-2-[1,5-二(二苯甲氧基)-4-吡啶酮-2-基甲氧基亞胺]乙酸的合成工藝改進
2016-02-25 05:47:36陶勻亮楊玉社汪海東
合成化學 2016年1期
陶勻亮, 楊玉社, 汪海東,3*
(1.常州大學 石油化工學院,江蘇 常州 213164; 2. 中國科學院 上海藥物研究所,上海 201203;
3.嘉興學院 生物與化學工程學院,浙江 嘉興 314000)
?

(Z)-2-(2-三苯甲基氨基噻唑-4-基)-2-[1,5-二(二苯甲氧基)-4-吡啶酮-2-基甲氧基亞胺]乙酸的合成工藝改進
陶勻亮1, 楊玉社2, 汪海東1,3*
(1.常州大學 石油化工學院,江蘇 常州213164; 2. 中國科學院 上海藥物研究所,上海201203;
3.嘉興學院 生物與化學工程學院,浙江 嘉興314000)
摘要:以5-羥基-2-羥甲基-4-吡喃酮為原料,經(jīng)羥基保護、邁克爾加成、親核取代、Mitsunobu反應、肼解及縮合等反應合成了BAL30072關(guān)鍵中間體——(Z)-2-(2-三苯甲基氨基噻唑-4-基)-2-(1,5-二(二苯甲氧基)-4-吡啶酮-2-基甲氧基亞胺)乙酸, 總收率23%,其結(jié)構(gòu)經(jīng)1H NMR和MS確證。該合成工藝已放大到公斤級規(guī)模。 2. 文稿內(nèi)容應按如下順序安排:中文和英文標題、作者姓名與單位、郵政編碼、
關(guān)鍵詞:曲酸; BAL30072; 革蘭氏陰性菌; 中間體; 合成; 工藝改進 (3~8個),正文(圖題和表題要求中文和英文對照)、
BAL30072是由巴塞利亞制藥公司(Basilea)研發(fā)的一種含鐵載體的新型單環(huán)β-內(nèi)酰胺抗生素,正處于Ⅰ期臨床試驗,其獨特的抗菌機制使其對多藥耐藥的鮑曼不動桿菌、大腸桿菌、綠膿桿菌及陰溝桿菌等革蘭氏陰性菌均十分有效[1-3]。是目前全球少數(shù)幾個進入臨床研究的具有抗多藥耐藥革蘭氏陰性菌活性的候選藥物之一,有望成為
治療由多藥耐藥革蘭氏陰性菌引起的感染性疾病的一線用藥。(Z)-2-(2-三苯甲基氨基噻唑-4-基)-2-[1,5-二(二苯甲氧基)-4-吡啶酮-2-基甲氧基亞胺]乙酸(1)是合成BAL30072的關(guān)鍵中間體,1再經(jīng)縮合和脫保護即可獲得BAL30072[4]。
到目前為止,合成1的路線鮮有報道。2008年Malcolm等[4]以5-羥基-2-羥甲基-4-吡喃酮(2)
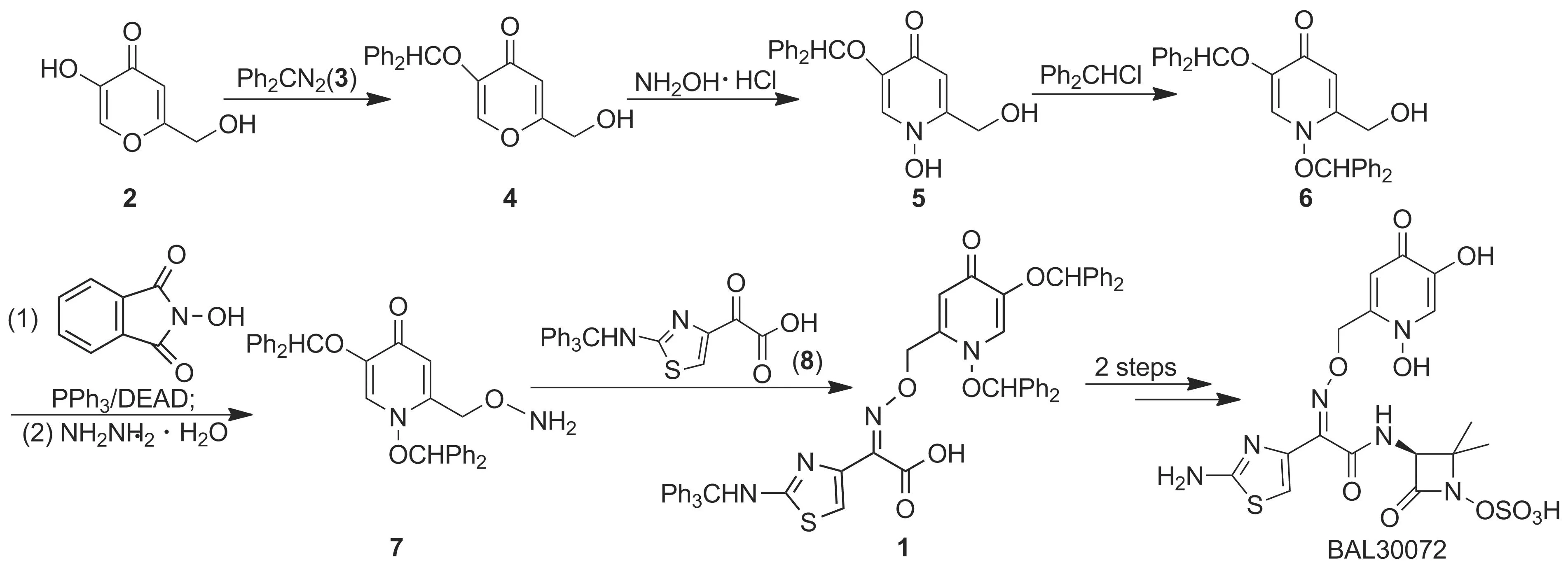
Scheme 1
為起始原料合成1。該合成方法使用高毒性溶劑苯,收率較低,后處理復雜,不利于工業(yè)化生產(chǎn)。
為了快速大量制備BAL30072,本文對上述合成路線進行了深入研究和優(yōu)化。以2為原料,經(jīng)酚羥基保護、邁克爾加成、親核取代、Mitsunobu反應及水合肼肼解制得2-氨基氧基甲基-1,5-二(二苯甲氧基)-4-吡啶酮(7); 7與2-三苯甲基氨基噻唑-4-基乙醛酸(8)經(jīng)縮合反應合成1(Scheme 1),總收率23%,其結(jié)構(gòu)經(jīng)1H NMR和MS確證。
該方法避免了高毒性溶劑苯的使用,減少了原料用量,簡化了后處理操作,總收率從10%提高至23%,適合工業(yè)化生產(chǎn)。目前,該合成工藝已放大到公斤級規(guī)模。
1實驗部分
1.1 試劑與儀器
X-4型數(shù)字顯微熔點儀(溫度未校正);Varian-400 MHz型核磁共振儀(CDCl3為溶劑,TMS為內(nèi)標);Trace DSQ FINNIGSN型質(zhì)譜儀;Agilent 1100型高效液相色譜儀[色譜柱:LATISIL ODS C18柱(4.6 mm×250 mm, 5 μm);流動相:V(乙腈) ∶V(0.3%三氟乙酸)=(80 ∶20);檢測波長210 nm;柱溫25 ℃]。
二苯基重氮甲烷(3)[6]和8[7]按文獻方法合成;其余所用試劑均為分析純。
1.2 合成
(1) 5-二苯甲氧基-2-羥甲基-4-吡喃酮(4)的合成
將2 100.0 g(0.7 mol)溶于乙醇(1 L)中,攪拌下加入3 135.0 g(0.7 mol),于40 ℃反應17 h(TLC監(jiān)測)。冷卻至室溫,減壓濃縮,殘余物依次用石油醚和水洗滌,于40 ℃真空干燥得白色固體4 168.4 g,收率77.6%, m.p.126~128 ℃;1H NMRδ: 7.45(s, 1H), 7.41~7.28(m, 10H), 6.48(s, 1H), 6.34 (s, 1H), 4.39(d,J=6.5 Hz, 2H), 2.88(t,J=6.6 Hz, 1H); ESI-MSm/z: 331.0{[M+Na]+}。
(2) 1-羥基-2-羥甲基-5-二苯甲氧基-4-吡啶酮(5)的合成
將4 150.0 g(0.486 mol)加入乙醇600 mL和水600 mL的混合溶液中,攪拌下加入鹽酸羥胺236.4 g(3.402 mol)和三水合乙酸鈉462.9 g,于60 ℃反應18 h(TLC監(jiān)測)。冷卻至室溫,抽濾,濾餅分別用水和乙醇洗滌,于40 ℃真空干燥得白色固體5 69.2 g,收率44%, m.p.222~224 ℃;1H NMR(DMSO-d6)δ: 10.71(s, 1H), 7.97(s, 1H), 7.55~7.23(m, 10H), 6.94(s, 1H), 6.65(s, 1H), 5.49(d,J=6.8 Hz, 1H), 4.39(d,J=3.2 Hz, 2H); ESI-MSm/z: 323.9{[M+H]+}。
(3) 1,5-二(二苯甲氧基)-2-羥甲基-4-吡啶酮(6)的合成
將5 60.0 g(0.186 mol)懸浮于二甲基亞砜600 mL中,攪拌下升溫至110 ℃使其溶解,冷卻至室溫,依次加入碳酸鉀38.5 g,碘化鈉41.8 g和二苯基氯甲烷51 mL (0.279 mol),于室溫反應17 h(TLC監(jiān)測)。倒入2 L冰水中,析出固體,過濾,濾餅用乙酸乙酯和石油醚混合溶液(V/V=2/1)洗滌,于40 ℃真空干燥得灰白色固體6 90.6 g,收率99.7%, m.p.123~125 ℃;1H NMR (DMSO-d6)δ: 7.60(s, 1H), 7.41~7.22(m, 20H), 6.41(s, 1H), 6.33(s, 1H), 6.02(s, 1H), 5.49(t,J=6.0 Hz, 1H), 4.11(d,J=5.8 Hz, 2H); ESI-MSm/z: 489.9{[M+H]+}。
(4) 7的合成
將6 80.0 g (0.163 mol)溶于二甲基亞砜300 mL和四氫呋喃500 mL中,依次加入N-羥基鄰苯二甲酰亞胺32.0 g和三苯基膦64.0 g,冰浴冷卻下,緩慢滴加偶氮二甲酸二乙酯(DEAD)38.4 mL,滴畢,于室溫反應1 h(TLC監(jiān)測)。加水200 mL,用乙酸乙酯(4×100 mL)萃取,合并萃取液,分別用飽和碳酸鉀溶液(4×100 mL)、水和飽和氯化鈉溶液洗滌,無水硫酸鈉干燥,減壓蒸除乙酸乙酯得油狀物A。
將A溶于乙醇500 mL中,加入85%水合肼8 mL (0.163 mol),攪拌下回流(75 ℃)反應2 h(TLC監(jiān)測)。冷卻至室溫,減壓蒸干,加入二氯甲烷300 mL,攪拌,抽濾,濾液分別用水(3×100 mL)和飽和氯化鈉溶液洗滌,無水硫酸鈉干燥,減壓蒸除溶劑,用二氯甲烷和石油醚重結(jié)晶得白色固體7 65.0 g,收率78.8%, m.p.141~143 ℃;1H NMRδ: 7.42~ 7.36(m, 5H), 7.33~7.27(m, 10H), 7.23~7.19(m, 5H), 6.76(s, 1H), 6.38(s, 1H), 6.07(s, 1H), 5.92(s, 1H), 4.32(s, 2H); ESI-MSm/z: 527.0{[M+Na]+}。
(5) 1的合成
將7 60.0 g (0.119 mol)溶于乙醇400 mL和二氯甲烷200 mL中,加入8 49.3 g(0.119 mol),攪拌下于室溫反應10 h(TLC監(jiān)測)。過濾,濾餅用二氯甲烷/石油醚(V/V=1/1)洗滌,于40 ℃真空干燥得白色固體1 91.1 g,收率85%, m.p.162~164 ℃,純度99.1%;1H NMR(DMSO-d6)δ: 8.84(s, 1H), 7.63 (s, 1H), 7.39~7.20(m, 35H), 6.85(s, 1H), 6.35(s, 1H), 6.32(s, 1H), 5.95(s, 1H), 4.72(s, 2H); ESI-MSm/z: 900.9{[M+H]+}; HR-ESI-MSm/z: Calcd for C56H44N4O6S{[M+Na]+}923.287 9, found 923.286 3。
2結(jié)果與討論
2.1 合成
(1) 4的合成
在4的合成中,文獻[5]方法采用n(2) ∶n(3)為1.0 ∶1.5進行投料,反應后剩余大量3,濃縮至干后得油狀物,需要用高毒試劑苯結(jié)晶,再用石油醚和水攪洗。本文考察了2和3的投料比對反應后處理的影響。實驗結(jié)果表明:當n(2)∶n(3)=1 ∶1時,3反應完全后,直接減壓蒸除溶劑析出固體,用石油醚和水洗滌即可高收率地制得4,無需用苯純化結(jié)晶。該方法簡化了操作并避免了高毒溶劑的使用,同時收率從56.8%提高至77.6%。
(2) 5的合成
在5的合成中,文獻[5]方法采用A=n(4) ∶n(鹽酸羥胺) ∶n(三水合乙酸鈉)=1 ∶10 ∶10。實驗發(fā)現(xiàn)當逐步減少鹽酸羥胺和三水合乙酸鈉的用量,A=1 ∶7 ∶7時也能獲得相同收率(44%);繼續(xù)減少鹽酸羥胺和三水合乙酸鈉的用量,當A=1 ∶6 ∶6時,收率降低為40%。因此選擇A=1 ∶7 ∶7為較佳投料比。既減少了原料的使用量,節(jié)省了資源,也降低了污染排放量。
(3) 7的合成
在7的合成中,文獻[5]方法先通過6與N-羥基鄰苯二甲酰亞胺進行Mitsunobu反應,反應后會產(chǎn)生大量三苯氧膦,且三苯氧膦和產(chǎn)物的極性非常相近,導致難以重結(jié)晶,需要通過柱色譜分離純化后再用水合肼肼解得到。由于需要柱色譜純化,使得該路線不利于工業(yè)化生產(chǎn),為大量制得BAL30072用于后續(xù)的研究造成困難。本文在Mitsunobu反應過程中嚴格控制原料中水分,使其盡可能保持干燥,同時細化了后處理方式:用乙酸乙酯萃取后,有機相用飽和碳酸鉀溶液洗去殘余的N-羥基鄰苯二甲酰亞胺,減壓濃縮后直接肼解,得到比三苯氧膦極性大的產(chǎn)物,濃縮后通過二氯甲烷和石油醚重結(jié)晶即可高收率地獲得7,從而解決了Mitsunobu反應帶來的后處理需要柱色譜純化的問題。
3結(jié)論
以5-羥基-2-羥甲基-4-吡喃酮為原料,經(jīng)6步反應合成了BAL30072關(guān)鍵中間體——(Z)-2-(2-三苯甲基氨基噻唑-4-基)-2-[1,5-二(二苯甲氧基)-4-吡啶酮-2-基甲氧基亞胺]乙酸,總收率23%。該工藝合成路線條件溫和,無需柱色譜純化,操作簡便,適合工業(yè)化生產(chǎn)。同時該合成方法為工業(yè)中應對使用Mitsunobu反應帶來的后處理困難提供了新的思路和方法。
參考文獻4. 文稿正文中所引應是作者親自閱讀過、最主要且發(fā)表在正式出版物上的。待發(fā)表的文獻及私人通信,不作為參考文獻著錄。正文中所引參考文獻的編號應同文末的文獻序號一致并附上方括號置于引用處右上角。日文文獻應在刊名后注明“(日)”字樣。參考文獻書寫格式如下: 。文后請附第一
[1]Mushtaq S, Warner M, Livermore D. Activity of the siderophore sulfactam BAL30072 against multiresistant non-fermenters[J]. J Antimicrob Chemother,2010,65:266-270.
[2]Mollmann U, Heinisch L, Bauernfeind A,etal. Siderophores as drug delivery agents:Application of the “Trojan Horse” strategy[J]. Biometals,2009,22:615-624.
[3]Malcolm G, Clothilde D, Eric D.InVitroproperties of BAL30072 a novel siderophor sulfactam with activity against multiresistant gram negative bacilli[J]. Antimicrobial Agents and Chemotherapy,2010,54(6):2291-2302.
[4]Malcolm G, Eric D. Combination medicaments for treating bacterial infections:WO 200 811 681[P].2008.
[5]Yoshiyuki Z, Nobuo I. Cephalosporin compounds pro-cesses for their preparation and antibacterial agents:US 4 883 879A[P].1989.
[6]呂茜茜,周曉靚,王榮先. 二苯基重氮甲烷的合成工藝改進[J].化學試劑,2008,30(2):147.
[7]Glinka T, Hecker S, Rodny D. Oxamazin antibiotics:WO 2 014 164 526[P].2014.

《合成化學》征稿簡則節(jié)選
一. 來稿要求及注意事項
1. 文稿應論點明確、文字精煉、數(shù)據(jù)可靠。
3. 文稿中的縮略詞及代號應在文中第一次出現(xiàn)處加括號寫明整個詞組或注明其含義。
(1) 期刊:[序號]作者.文獻題名[J].刊名,年,卷(期):起頁碼-止頁碼.
(2) 專著:[序號]作者.書名[M].其他責任者(如編者、譯者).版次(第1版不標注),卷(冊),出版地:出版社名,出版年:頁碼.
(3) 專利:[序號]專利申請者.題名:[P].專利國別(或地區(qū)),專利號,年份.
(4) 學位論文:[序號]作者.文獻題名[D].學校地點:授予學位單位全稱,年份.
(5) 會議論文:[序號]編者.會議文集名稱[C].會議地點,年份.出版地:出版者,出版年:頁碼.
(6) 標準:[序號]標準編號,標準名稱[S].
二. 其它
1. 來稿“文責自負”,編輯部對稿件有刪改權(quán)和出版使用權(quán)。請勿一稿兩投。稿件發(fā)表周期一般在12個月以內(nèi),若作者要求提前發(fā)表,請在收到審稿結(jié)果時說明。校清樣時,全體作者與本刊簽署《論文著作權(quán)轉(zhuǎn)讓書》。來稿一經(jīng)發(fā)表,即贈送該期期刊兩冊,并付一次性稿酬。
2. 聯(lián)系方式: 四川省成都市武侯區(qū)人民南路四段九號,中國科學院成都有機化學有限公司,《合成化學》編輯部,郵編: 610041,電話: 028-85255007,網(wǎng)站: http:∥hchxcioc.com,郵箱: hchx@cioc.ac.cn
·研究簡報·
通信聯(lián)系人: 汪海東,教授,碩士生導師, Tel. 0763-83646686, E-mail: whdoctor@163.com
Process Improvement on the Synthesis of (Z)-2-(2-tritylaminothiazol-
4-yl)-2-(1,5-dibenzhydryloxy-4-pyridon-2-ylmethoxyimino)acetic Acid
TAO Yun-liang1, YANG Yu-she2, WANG Hai-dong1,3*
(1. School of Petrochemical Engineering, Changzhou University, Changzhou 213164, China;
2. Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China;
3. College of Biological, Chemical Sciences and Engineering, Jiaxing University, Jiaxing 314000, China)
Abstract:The key intermediate of BAL30072, (Z)-2-(2-tritylaminothiazol-4-yl)-2-[(1,5-dibenz-hydryloxy)-4-pyridon-2-ylmethoxyimino]acetic acid with overall yield of 23%, was synthesized by protection of phenolic hydroxyl group, Michael addition, nucleophilic substitution, Mitsunobu reaction, hydrazinolysis, condensation reaction and so on, using kojic acid as the starting material. The structure was confirmed by1H NMR and MS.
Keywords:kojic acid; BAL30072; gram-negative bacteria; intermediate; synthesis; process improvement
中圖分類號:O626.41; R914.5 、中文和英文(包括研究目的、研究背景、研究方法、結(jié)果與討論等內(nèi)容)及
文獻標志碼:A
DOI:10.15952/j.cnki.cjsc.1005-1511.2016.01.15044
作者簡介:陶勻亮(1990-),男,漢族,浙江溫嶺人,碩士研究生,主要從事藥物及中間體的合成研究。 E-mail: tyliang322@163.com (姓名、出生年、性別、民族、籍貫、職稱、學位、簡歷或研究方向);基金資助情況(項目批準號);通訊聯(lián)系人的姓名、職稱、聯(lián)系電話、E-mail。
收稿日期:2015-03-05;
修訂日期:2015-10-07
