PBT/增塑劑共混物相容性的介觀動力學模擬
2015-12-31 11:58:40李苗苗孫慶鋒干效東郭效德
上海航天 2015年4期
關鍵詞:體系
李苗苗,孫慶鋒,干效東,郭效德
(1.上海航天動力技術研究所,浙江 湖州 313000;2.南京理工大學 化工學院,江蘇 南京 210094)
0 引言
PBT是一種含能粘合劑,具高能、高燃速、低特征信號等優點。采用該粘合劑的推進劑在滿足安全性能的前提下,可有效提高推進劑的能量和密度,是當前高能推進劑發展的主要方向[1]。為改善推進劑的力學性能,增加PBT柔韌性并使之易于加工,需加入功能組分增塑劑,增塑劑與PBT須具有良好的相容性,能充分溶解在粘合劑網絡中。推進劑中高分子共混物的相容性可通過實驗方法表征,其中較簡便的是溶度參數法。有研究提出用內聚能密度的概念表征物質內分子間的吸引力即溶度參數,但聚合物的溶度參數只能由溶脹法、特性黏數法、滲透壓法等間接獲取,過程費時費力或存在某些局限性。計算機模擬技術克服了上述不足,近年來已有越來越多的研究應用分子模擬和介觀模擬方法研究推進劑配方中各組分的相容性[2-6]。但之前的研究多集中于 HTPB推進劑,利用分子動力學和介觀動力學方法研究PBT與增塑劑共混體系的相容性少見報道。本文用 MD,MesoDyn對 PBT/A3 和 PBT/A3/GAPA兩種體系進行了研究,對體系的溶度參數、共混物分子間的Flory-Huggins作用參數和介觀形貌進行模擬計算,預測共混物的相容性。
1 模型構建與模擬方法
1.1 分子模型構建和MD模擬細節
用Materials Studio(MS)軟件包中Visualizer模塊依據表1建立PBT,A3,GAPA的相應分子模型[7]。其中:取PBT硬段鏈節數m=21,軟段鏈節數n=21;GAPA鏈節數m=5。將所得的高聚物模型,用Discover模塊,以Compass力場進行2.5ns MD模擬,獲得的最終結構視為高聚物鏈的平衡構象。在溫度298K,壓力1.01×105Pa條件下,按相應的密度分別構建PBT,A1,A2,GAPA的無定型分子模型如圖1所示。用Smart Minimization法對構建的無定型分子模型進行結構優化,再進行每隔50K,從300K升溫到600K再降溫到300K的五個循環的退火處理(該過程可基本消除構建的模型中產生的局部不合理結構,為進行下一步的MD模擬提供較合理的平衡幾何構象)。
結構優化后的模型進行MD模擬,用Andersen控溫方法、Berendsen控壓方法,各分子起始速度按Maxwell分布取樣,Velocity Verlet算法進行求解,范德華和靜電作用,分別用Atom-based,Ewald法,非鍵截取半徑0.95nm,樣條寬度0.1nm,緩沖寬度0.05nm,時間步長1fs,進行200ps NPT系綜的MD模擬,后100ps用于分析性能,力場選擇Compass力場[8-13]。
1.2 Mesodyn模擬細節
在Mesodyn介觀模擬中,為表征體系的化學性質,需確定如下兩類重要參數。
a)表示各重復單元的高斯鏈
在高斯鏈中,所有珠子有相同的體積,原始體系的粗化程度影響高斯鏈的拓撲結構。由高分子鏈粗粒化后包含的珠子數
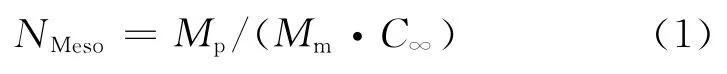
可得模擬分子的高斯鏈結構。此處:Mp為聚合物分子量;Mm為重復單元分子量;C∞為高分子鏈的極限特征比,為高分子鏈的固有特性,可表征高分子鏈的柔順性。PBT的Mp=5 057,Mm=240;GAPA的Mp=495,Mm=99。用 MS軟件Synthia模塊中的QSAR方法求得PBT,GAPA的C∞分別為5.86,5.3,這樣分別用7個珠子和1個珠子替代PBT,GAPA分子鏈。另外,分別用1個珠子替代A1,A2分子。
b)不同珠子間的相互作用參數
先求解PBT與增塑劑的Flory-Huggins參數χ,χ乘以RT即為MesoDyn模擬中輸入的相互作用參數值。此處:R為理想氣體常數;T為溫度。模擬中所用的盒子尺寸32.0nm×32.0nm×32.0nm,珠子的擴散系數1.0×10-7cm2/s,設體系的噪聲參數為75,可壓縮參數為10。考慮體系的平衡時間,取模擬步長50ns,模擬總步數20 000步,總模擬時間為1 000μs。
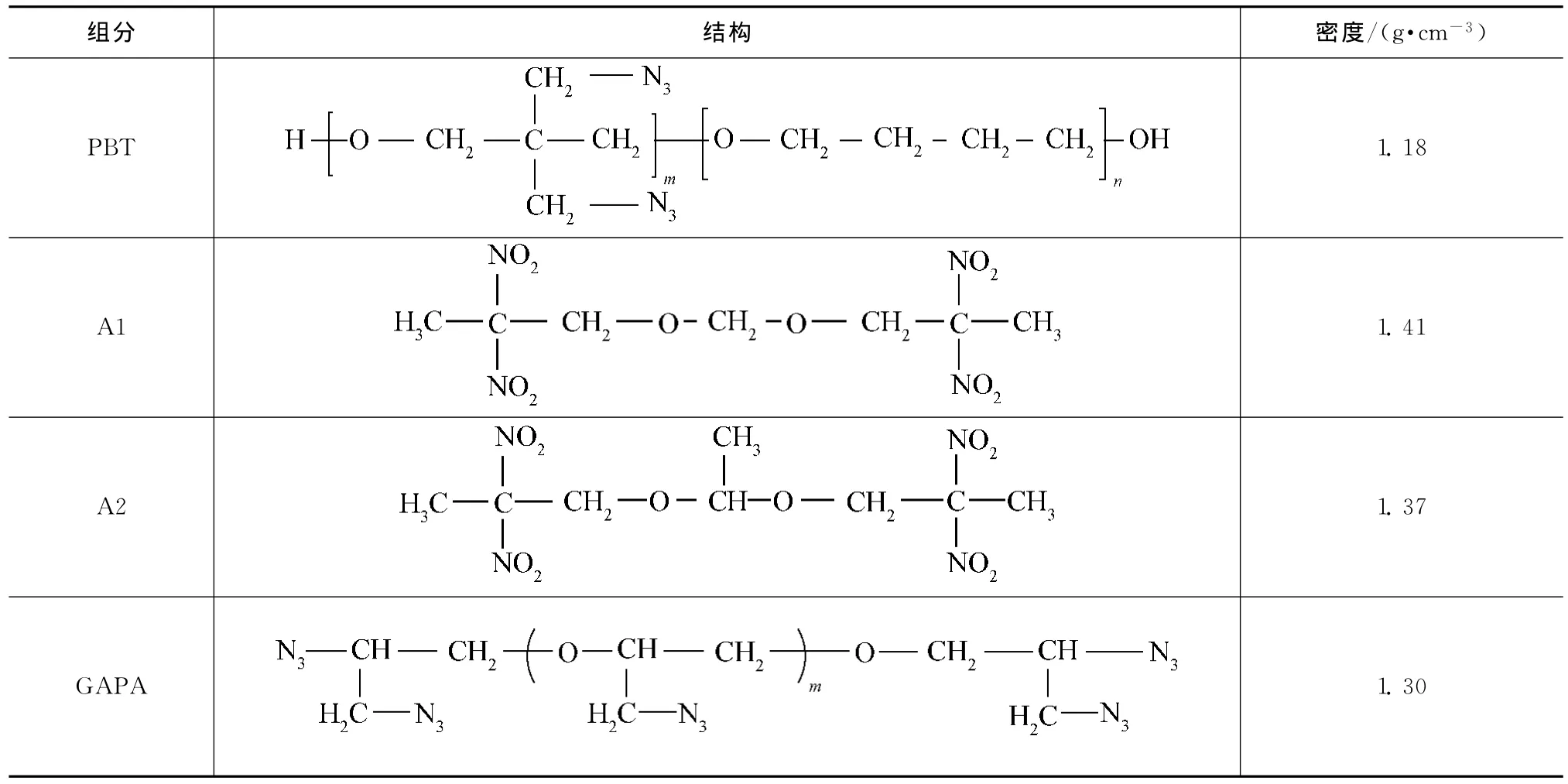
表1 PBT,A1,A2,GAPA結構與性質Tab.1 Structure and physical properties of PBT,A1,A2and GAPA
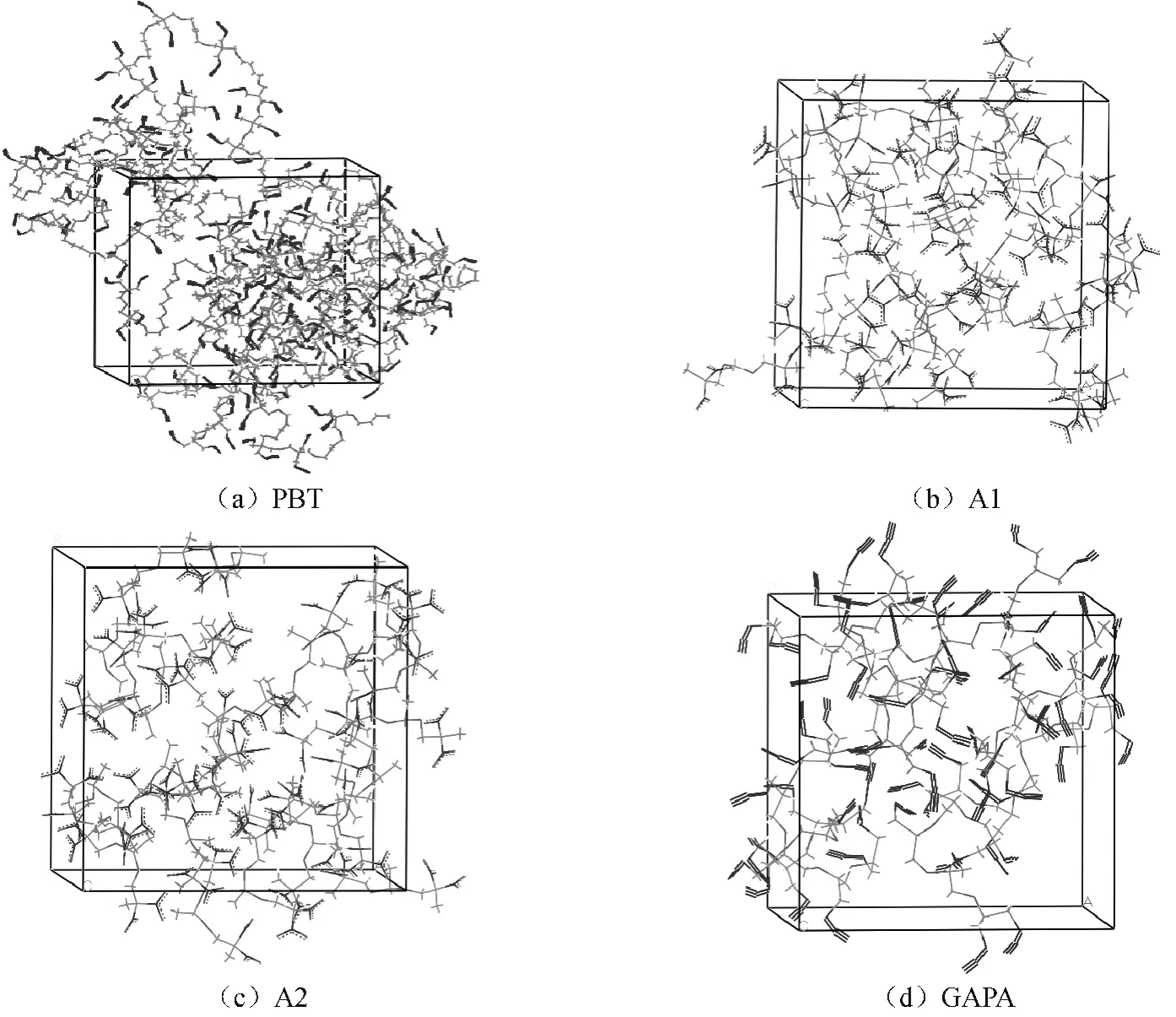
圖1 純物質PBT,A1,A2,GAPA的無定形分子模型Fig.1 Amorphous cell structure of pure substances PBT,A1,A2and GAPA
2 結果與討論
2.1 溶解度參數與Flory-Huggins參數
因粘合劑與增塑劑分子結構的差異,兩者存在相容性問題,粘合劑與增塑劑相容性直接影響推進劑性能。實際應用中,多用溶解度參數評價粘合劑與增塑劑的相容性,兩者的溶解度參數越接近,相容性就越好。一般認為,只要粘合劑與增塑劑的溶解度參數之差小于1.3~2.1(J/cm3)1/2,兩者就具有良好的相容性[5]。溫度298K 時,PBT,A1,A2,GAPA的溶度參數計算值分別為20.36,22.89,22.33,21.24 (J/cm3)1/2,參數差值較小,表明它們在室溫下有良好的相容性。
在MesoDyn模擬中,珠子間的相互作用參數可用不同珠子間的χ表示,有
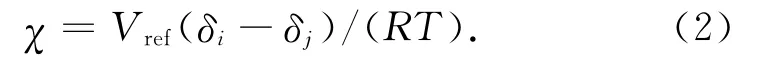
式中:Vref為 重 復 單 元 參 考 體 積;δi,δj分 別 為 PBT和增塑劑的溶度參數。用式(2)求得χ后乘以RT即為Mesodyn模擬中輸入的相互作用參數值ν-1εij。
2.2 PBT/A3共混物體系相容性
研究了在溫度298K條件下,增塑比(mA3/mPBT)分別為1.0,1.2,1.4,1.6,1.8,2.0,2.5,3.0時共混物的介觀結構。定義有序度參數
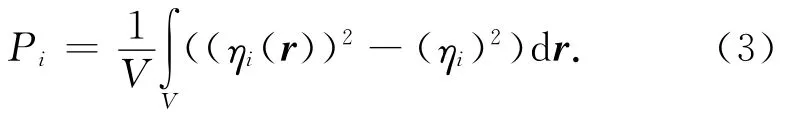
式中:V為格子的體積;ηi為組分i的無量綱密度(體積分數);r為位置矢量。Pi表示體系與均相分布的差異程度,反映了體系的相分離過程,是體系相分離和各組分相容程度的綜合體系,其值越大,相容性越差。不同增塑比PBT/A3共混物中PBT的有序度參數如圖2所示。由圖2可知:當增塑比為1時,有序度參數較大,相容性較差,可能會產生相分離;當增塑比大于1.2時,體系的有序度參數較低;增塑比為1.4時,有序度參數達到最低點,此時體系熵值較小,相容性最好。
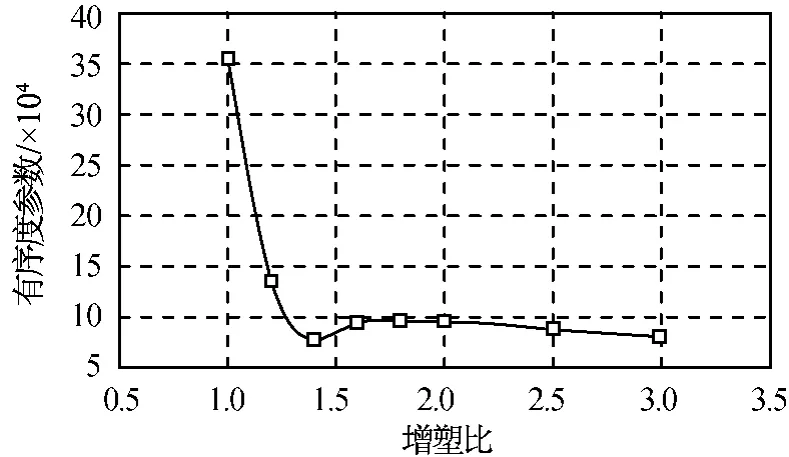
圖2 不同增塑比PBT/A3共混物有序度參數Fig.2 Mesodynamic order parameter for PBT/A3 blends with different plasticizing ratio
增塑比為1.0,1.2,1.4時共混物介觀動力學模擬的等密度圖如圖3所示。由圖3可知:增塑比對共混體系的介觀形貌影響較大,當增塑比為1.0時,體系發生了明顯的層狀相分離;當增塑比大于1.2時,增塑劑可均勻分散在PBT粘合劑中,形成均相體系,其中增塑比為1.4時分散效果最好,與有序度參數的變化趨勢一致。
2.3 PBT/A3/GAPA共混物體系相容性
GAPA是由加拿大國防部AMPLEMAN和美國Rockwell公司WILSON制備出的一類疊氮基化合物,其端疊氮基熱分解是獨立進行且先于主鏈上的疊氮基分解,故它不但能增加配方能量,還能起到加速推進劑分解的作用,同時機械感度又較硝酸酯低。本文對PBT/A3體系添加GAPA后的相容性進行研究,以增塑比為1.2的PBT/A3/GAPA共混體系為例,設計的計算體系分別為PBT/A3/GAPA=10/0/12,10/2/10,10/4/8,10/6/6,10/8/4,10/10/2,10/12/0,對應 A3的質量分數分別為0%,9.09%,18.18%,27.27%,36.36%,45.45%,54.55%。用 Mesodyn介觀模擬計算,共混物中PBT有序度參數如圖4所示。由圖可知:除10/8/4體系的有序度參數較大外,其余配方的有序度參數相對較小。

圖3 不同增塑比PBT/A3共混物等密度圖Fig.3 Morphologies of PBT/A3blends at different plasticizing ratio
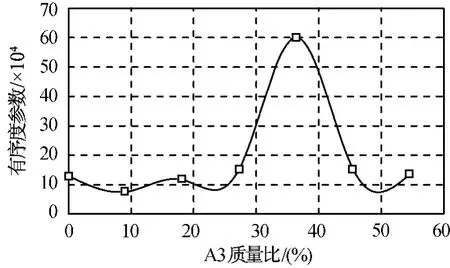
圖4 不同配比PBT/A3/GAPA共混物有序度參數Fig.4 Mesodynamic order parameter for PBT/A3/GAPA blends with different A3content
配比為10/0/12,10/2/10,10/8/4的共混物介觀動力學模擬的等密度圖如圖5所示。由圖可知:配方變化對共混體系的介觀形貌影響較大,10/0/12,10/2/10體系中各組分分布相對較均勻,而10/8/4體系發生了明顯的層狀相分離,與圖4中有序度參數的變化趨勢一致。
3 結論
本文用MD和MesoDyn模擬了PBT,A1,A2,GAPA物質的溶解度參數,比較溶解度參數值的大小可預測其在室溫下有無良好的相容性。對不同增塑比的PBT/A3共混物介觀形貌與動力學演變過程研究發現增塑比為1.4時體系的相容性最好。對增塑比為1.2的PBT/A3/GAPA共混物介觀形貌與動力學演變過程研究發現PBT/A3/GAPA配比為10/2/10體系的相容性最好。本文研究可為PBT推進劑配方設計提供理論參考。

圖5 不同配比的PBT/A3/GAPA共混物的等密度圖Fig.5 Morphologies of PBT/A3/GAPA blends at different mixure ratio
[1] 李 祎,盧興福,吳戰鵬.BAMO/THF疊氮低特征信號推進劑研究[A].2002年火炸藥技術及鈍感彈藥學術研討會論文集[C].珠海:中國工程物理研究院化工材料研究所,2002:95-101.
[2] FRAAIJE J G E M,VAN VLIMMEREN B A C,MAURITS N M,et al.The dynamic mean-field density functional method and its applieaction to the mesoscopic dynamics of quenched block copolymer melts[J].J Chem Phys,1997,106:4260-4269.
[3] GROOT R D,MADDEN T J.Dynamic simulation of diblock copolymer microphase separation[J].J Chem Phys,1998,108:8713-8724.
[4] 趙貴哲,馮益柏,付一政,等.端羥基聚丁二烯/增塑劑共混物相容性的分子動力學模擬和介觀模擬[J].化學學報,2009,67(19):2233-2238.
[5] 趙樹森,付一政,梁曉艷,等.HTPB/增塑劑共混物的介觀動力學模擬[J].高分子材料科學與工程,2011,27(5):186-190.
[6] 焦東明,楊月誠,強洪夫,等.HTPB固體推進劑增塑劑選取分子模擬研究[J].化學研究與應用,2009,21(6):805-809.
[7] ACCELRYS Inc.CA materials studio 3.0.1[M].San Diego:Accelrys Inc,2004.
[8] ANDERSEN H C.Molecular dynamics simulations at constant pressure and/or temperature[J].J Chem Phys,1980,72(4):2384-2393.
[9] BERENDSEN H J C,POSTMA J P M,VAN GUNSTEREN W F,et al.Molecular dynamics with coupling to an external bath[J].J Chem Phys,1984,81(8):3684-3690.
[10] ALLEN M P,TILDESLEY D J.Computer simulation of liquids[M].Oxford:Clarendon Press,1987:78-80.
[11] KARASAWA N, GODDARD W.Force fields,structures,and properties of poly(vinylidene fluoride)crystals[J].Macromolecules,1992,25 (26):7268-7281.
[12] EWALD P P.Evaluation of optical and electrostatic lattice potentials[J].Annalen der Physik,1921,369(3):253-287.
[13] SUN H.COMPASS:an ab initio force-field optimized for condensed-phase applications overview with details on alkane and benzene compounds[J].J Chem Phys B,1998,102(38):7338-7364.
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
