高效液相色譜梯度洗脫法測定水楊酸復合洗劑中的有關物質
2015-06-24 14:29:48郭旭光鄭子棟郭毅
醫藥導報 2015年5期
郭旭光,鄭子棟,郭毅
(河南省食品藥品檢驗所,鄭州 450003)
高效液相色譜梯度洗脫法測定水楊酸復合洗劑中的有關物質
郭旭光,鄭子棟,郭毅
(河南省食品藥品檢驗所,鄭州 450003)
目的 建立梯度洗脫的高效液相色譜(HPLC)法測定水楊酸復合洗劑中的有關物質。方法 采用Agela C18(250 mm×4.6 mm,5 μm),以0.5%磷酸溶液(A)、甲醇(B)為流動相,梯度洗脫,流速為1.0 mL·min-1,檢測波長為270 nm,柱溫為35 ℃。結果 該方法可快速有效分離水楊酸復合洗劑中各有關物質。結論 該方法準確、靈敏、快速、專屬性強,可以用于控制該產品質量。
水楊酸復合洗劑;梯度洗脫;色譜法,高效液相
水楊酸復合洗劑具有止癢、殺菌的作用,用于真菌感染的手、足癬的治療,療效好,價格便宜。現行水楊酸復合洗劑的質量標準沒有對其有關物質進行檢定[1],筆者參考相關文獻采用高效液相色譜梯度洗脫法對水楊酸復合洗劑的有關物質進行了測定[2-10],本方法快速,簡便,準確,可以更好地控制該產品質量。
1 儀器與試藥
1.1 儀器 Agilent 1100型高效液相色譜儀(美國Agilent公司);G1314A VWD可變波長檢測器(美國Agilent公司);Agilent chem Station色譜工作站。
1.2 試藥 苯甲酸對照品(中國食品藥品檢定研究院,批號:100419-200301,含量:100%),水楊酸對照品(中國食品藥品檢定研究院,批號:100106-201104,含量:100%),4-羥基苯甲酸對照品(Acros公司,批號:A0282187,含量:99.8%),4-羥基異酞酸對照品(東京化成工業株式會社,批號:DLTTC,含量:99.4%),苯酚對照品(中國食品藥品檢定研究院,批號:100509-200902,含量:100%),水楊酸復合洗劑(市售,批號:130101,130102,130103),甲醇為色譜純,其他試劑均為分析純,水為重蒸水。
2 方法與結果
2.1 色譜條件 色譜柱:Agela C18(250 mm×4.6 mm,5 μm);流動相A為0.5%磷酸溶液,流動相B為甲醇,線性梯度洗脫程序見表1;柱溫:35 ℃;流速為1.0 mL·min-1,檢測波長為270 nm;進樣體積為10 μL。
2.2 系統適用性實驗 取苯甲酸、水楊酸、4-羥基苯甲酸、4-羥基異酞酸及苯酚對照品適量,置同一量瓶中,用0.5%磷酸溶液-甲醇(1:1)混合溶液溶解稀釋制成濃度為16 μg·mL-1的溶液,作為系統適用性溶液,按上述色譜條件測定。結果表明,相鄰峰的分離度均>1.5。
表1 梯度洗脫程序

%
2.3 專屬性實驗
破壞性實驗 儲備液的制備:精密稱取粉劑300 mg,置20 mL量瓶中,用0.5%磷酸溶液-甲醇(1:1)混合溶液溶解,稀釋至刻度,搖勻。
酸破壞:精密量取儲備液2 mL,置5 mL量瓶中,加1 mol·L-1鹽酸1 mL放置2 h后用1 mol·L-1氫氧化鈉調pH至中性,加0.5%磷酸溶液-甲醇(1:1)混合溶液稀釋至刻度,搖勻,用孔徑0.2 μm微孔濾膜濾過,取續濾液,按上述色譜條件測定。
堿破壞:精密量取儲備液2 mL,置5 mL量瓶中,加1 mol·L-1氫氧化鈉1 mL放置2 h后用1 mol·L-1鹽酸調pH至中性,加0.5%磷酸溶液-甲醇(1:1)混合溶液稀釋至刻度,搖勻,用孔徑0.2 μm微孔濾膜濾過,取續濾液,按上述色譜條件測定。
氧化破壞:取儲備液2 mL,置5 mL量瓶中,加30%過氧化氫1 mL,加0.5%磷酸溶液-甲醇(1:1)混合溶液稀釋至刻度,搖勻,放置0.5 h后,用孔徑0.2 μm微孔濾膜濾過,取續濾液,按上述色譜條件測定。
高溫破壞:取儲備液2 mL,置5 mL量瓶中,60 ℃水浴2 h,放至室溫,加0.5%磷酸溶液-甲醇(1:1)混合溶液稀釋至刻度,搖勻,用孔徑0.2 μm微孔濾膜濾過,取續濾液,按上述色譜條件測定。
光照破壞:取儲備液2 mL,置5 mL量瓶中,加0.5%磷酸溶液-甲醇(1:1)混合溶液稀釋至刻度,搖勻,于4 000 lx光照2 h,放至室溫,用孔徑0.2 μm微孔濾膜濾過,取續濾液,按上述色譜條件測定。
結果各破壞條件下的降解物與主峰分離均良好。
2.4 線性范圍考察 精密稱取苯甲酸、水楊酸、4-羥基苯甲酸、4-羥基異酞酸和苯酚對照品各20 mg置同一50 mL量瓶中,加0.5%磷酸溶液-甲醇(1:1)混合溶液溶解并稀釋至刻度,搖勻,精密量取5 mL,置于50 mL量瓶中,加0.5%磷酸溶液-甲醇(1:1)混合溶液稀釋至刻度,搖勻。精密量取0.5,2.0,3.0,5.0,7.0,9.0 mL分別置10 mL量瓶中,加0.5%磷酸溶液-甲醇(1:1)混合溶液稀釋至刻度,搖勻,按上述色譜條件測定,記錄色譜圖,以濃度C(μg·mL-1)對峰面積A進行線性回歸,分別得苯甲酸、水楊酸、4-羥基苯甲酸、4-羥基異酞酸和苯酚的線性回歸方程:
A=2.720 4C+0.320 1,R2=0.999 9(n=6);
A=3.479 5C+0.675 4,R2=0.999 6(n=6);
A=63.047C+6.185 5,R2=0.999 8(n=6);
A=37.58C-3.457 1,R2=0.999 9(n=6);
A=3.292 3C+0.459 6,R2=0.999 8(n=6)。
結果表明,苯甲酸、水楊酸、4-羥基苯甲酸、4-羥基異酞酸和苯酚分別在2.109~37.962 μg·mL-1、1.989~35.802 μg·mL-1、1.982~35.676 μg·mL-1、2.018~36.324 μg·mL-1和2.234~40.212 μg·mL-1范圍內,濃度與峰面積呈良好線性關系。
2.5 精密度實驗 取“2.2”項下的系統適用性實驗溶液,分別連續進樣6次,按“2.1”項色譜條件進行分析,記錄色譜圖,測得苯甲酸、水楊酸、4-羥基苯甲酸、4-羥基異酞酸和苯酚峰面積的RSD 分別為0.8%,0.5%,0.6%,0.5%和0.4%,結果表明此法精密度良好。
2.6 重復性實驗 精密稱取水楊酸復合洗劑(批號:130101)6份,制備供試品溶液,按上述色譜條件測定6份樣品有關物質總峰面積、雜質個數、單個雜質量、雜質總量。結果總峰面積平均值為14 536.5,RSD為0.9%;雜質個數均為5個;單個雜質量基本沒有變化;雜質總量平均值為0.55%,RSD為0.2%。
2.7 檢測限和定量限 以信噪比為3:1,測得苯甲酸、水楊酸、4-羥基苯甲酸、4-羥基異酞酸和苯酚檢測限分別為4.22,3.98,0.124,0.448和4.47 ng;定量限分別為12.78,12.06,0.37,1.49和13.45 ng。
2.8 溶液穩定性 選取2批樣品(批號:130101,130102)進行考察。結果顯示,供試品溶液在常溫下放置24 h,2批樣品的雜質峰數目、單個雜質的峰面積和雜質總量均無變化(RSD=0.04%),說明溶液在常溫下24 h內穩定。
2.9 耐用性 選用2根不同品牌的C18柱,Agela Venusil ASB C18(250 mm×4.6 mm,5 μm)和Agilent Eclipse XDB C18(250 mm×4.6 mm,5 μm),在2臺儀器(Agilent 1100型高效液相色譜儀和Waters 2695型高效液相色譜儀)上測定3批樣品的有關物質,發現不同色譜柱檢測到的雜質數目、出峰順序、分離效果和峰面積均無明顯變化。
2.10 樣品測定 取本品適量,精密稱定,用0.5%磷酸溶液:甲醇(1:1)混合溶液定量稀釋制成每毫升中含水楊酸約2 mg的溶液,搖勻,用孔徑0.2 μm微孔濾膜濾過,取續濾液作為供試品溶液;精密量取供試品溶液1 mL,置于100 mL量瓶中,0.5%磷酸溶液:甲醇(1:1)混合溶液稀釋至刻度,搖勻,經孔徑0.2 μm濾膜濾過,將濾液作為對照溶液。按上述色譜條件測定,分別取對照溶液及供試品溶液各10 μL注入液相色譜儀,記錄色譜圖,供試品溶液色譜圖中如有雜質峰,單個雜質峰的峰面積不得大于對照溶液中水楊酸主峰面積的0.5倍;各雜質峰的面積之和不得大于對照溶液中水楊酸峰面積的3倍。用自身對照法計算各雜質的含量,3批有關物質均符合規定,見表2。
3 討論
筆者比較了4種不同流動相系統:①甲醇-水-冰醋酸(60:40:1)[2];②甲醇-水-冰醋酸(40:60:1)[3];③20 mmol·L-1磷酸二氫鉀溶液(用20%磷酸調節pH為2.7)-甲醇(40:60);④以0.5%磷酸溶液
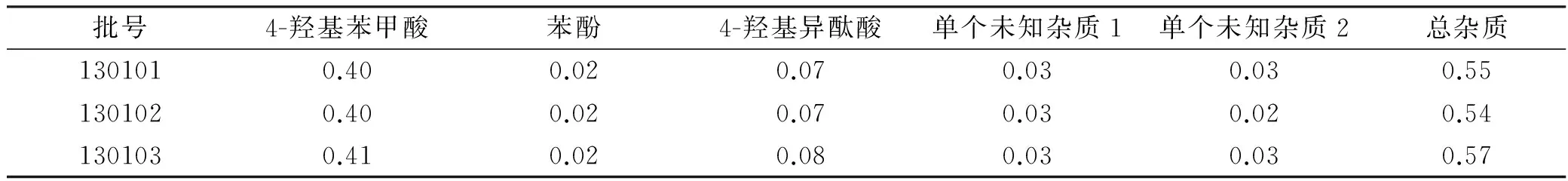
表2 有關物質測定結果 %
(A)、甲醇(B)為流動相,梯度洗脫。結果流動相①、②和③部分色譜峰的分離度<1.5,并且基線較高且有漂移,半峰寬較大,峰拖尾因子不理想,水楊酸峰后出現一較大寬峰;采用流動相④梯度洗脫后基線漂移現象得到改善,水楊酸后的大寬峰得以改善,主峰及各雜質峰之間均分離良好;筆者嘗試用0.5%三氟乙酸溶液和0.5%醋酸溶液替代流動相④中的0.5%磷酸溶液進行梯度洗脫實驗,結果流動相A為0.5%三氟乙酸溶液時圖譜中各峰的分離度均>4.0,分離度較好且柱效高,但是基線噪音大;用0.5%醋酸溶液以同樣梯度洗脫,結果苯甲酸和水楊酸分離不完全。嘗試改變梯度程序,主峰保留時間、檢出雜質峰個數及分離度均有所不同,綜合考慮以上因素并結合柱效和拖尾因子,最終確定流動相④的梯度洗脫程序最優,筆者選擇流動相④梯度洗脫作為該產品流動相系統。
分別精密稱取兩份水楊酸復合洗劑樣品(批號:130101)分別用溶劑A[0.5%磷酸溶液-甲醇(1:1)混合溶液]和溶劑B(50%甲醇)稀釋制成每毫升含水楊酸約2 mg的溶液,按本文色譜條件進樣,結果溶劑B稀釋的樣品色譜圖中苯甲酸和水楊酸兩主峰分離不完全;用溶劑A稀釋后的色譜圖,主峰之間及與雜質峰均分離完好,故確定溶劑A為本文方法溶劑。
采用DAD檢測器對供試品溶液色譜圖中的各雜質峰和主峰的最大紫外吸收進行測定,經實驗圖譜中各峰的紫外吸收主要集中在254,270和230 nm附近,在這3個波長處,各雜質的吸收大小程度不同,分別對同一份供試品進行有關物質測定,結果表明在270 nm波長處單個雜質峰的面積百分比均較大,檢出的總雜質量最多,為了更多地檢出雜質,更有效地控制該產品質量,選擇270 nm為有關物質檢測波長。
本品的藥效成分是苯甲酸、水楊酸、硼酸和乳酸。苯甲酸和水楊酸具有紫外吸收,筆者依此并參考相關文獻建立了高效液相色譜梯度洗脫法測定其有關物質[2-10],因硼酸和乳酸沒有紫外吸收,其有關物質暫時還沒有檢測方法;4-羥基苯甲酸、4-羥基異酞酸和苯酚均為已知雜質,含量均<0.5%,其他雜質檢出量均非常微小,本研究共檢出兩個單個未知雜質,其結構和歸屬有待進一步研究,兩個未知單個雜質均暫以主成分水楊酸峰面積計算。參照《中華人民共和國藥典》[2]水楊酸原料標準中對雜質A(4-羥基苯甲酸)、雜質B(4-羥基異酞酸)、雜質C(苯酚)的限度規定,確定本品單個雜質的限度不超過0.5%,以更好地控制該產品質量。
破壞性實驗表明本品在強酸堿和氧化條件下均有降解,光破壞和高溫破壞幾乎沒有降解,說明儲存過程中要避免強酸堿條件并應密封保存,考慮到苯甲酸和水楊酸均為小分子物質,降解后可能沒有紫外吸收,所以有關物質和含量測定相結合以進行質量評價更有意義。
[1] 國家藥典委員會.國家藥品標準新藥轉正標準(第13冊)[S].北京:中國醫藥科技出版社,1998:75.
[2] 國家藥典委員會.中華人民共和國藥典(二部)[M].北京:中國醫藥科技出版社,2010:78-79.
[3] The United States Pharmacopeial Convention.USP34-NF29[M].Maryland:the United States Pharmacopeial Convention,2011:4194-4195.
[4] European Directorate for Quality Medicines.EP7.2[M].Strasbourg:European Directorate for Quality Medicines,2011:2884-2885.
[5] 黃潔,李鋒武,劉鵬鳴,等.HPLC法測定水楊酸原料藥的有關物質[J].藥物分析雜志,2011,31(11):2182-2184.
[6] 李慧敏.HPLC法檢測口服液類保健食品中苯甲酸和山梨酸的含量[J].中國藥事,2013,27(1):38-40.
[7] 章家偉,王輝,孫慶榮,等.RP-HPLC法同時測定苯甲酸原料藥中有關物質苯甲醛和甲苯的含量[J].中國藥房,2013,24(29):2754-2757.
[8] 何秀峰,夏鵬飛,王愛國,等.水楊酸甲酯乳糖苷有關物質測定方法的研究[J].中國新藥雜志,2013,22(11):1259-1262.
[9] 熊苗苗,汪秋蘭,施春陽,等.高效液相色譜法測定復方磷酸哌喹片的有關物質[J].醫藥導報,2013,32(1):75-77.
[10] 付桂英,李秀青,孫燕,等.HPLC法測定水楊酸軟膏中主藥及有關物質的含量[J].中國藥房,2009,20(16):1257-1258.
DOI 10.3870/yydb.2015.05.026
2014-02-21
2014-04-26
郭旭光(1972-),男,河南襄城人,主管藥師,碩士,主要研究方向:食品、藥品質量控制。電話:0371-63388290,E-mail:gxg0371@126.com。
R286;R927.2
B
1004-0781(2015)05-0664-03
