21例Crouzon綜合征頭顱三維CT分析
2015-04-14 02:59:07謝毓芝朱文靜
海南醫學 2015年2期
關鍵詞:融合
謝毓芝,林 濤,朱 凌,朱文靜
(1.上海交通大學醫學院附屬第九人民醫院放射科,上海 200011;2.上海浦東新區周浦醫院放射科,上海 201318)
21例Crouzon綜合征頭顱三維CT分析
謝毓芝1,林 濤2,朱 凌1,朱文靜1
(1.上海交通大學醫學院附屬第九人民醫院放射科,上海 200011;2.上海浦東新區周浦醫院放射科,上海 201318)
目的 探討Crouzon綜合征CT影像表現。方法回顧性分析21例Crouzon綜合征患者的頭顱三維CT影像資料,總結其異常CT表現。結果Crouzon綜合征頭顱三維影像表現包括顱骨、眼眶、面中部及頸椎的異常。顱骨:5例舟狀頭畸形,4例尖頭畸形,3例短頭畸形;15例顱骨內板“指壓跡”,12例垂體窩增大;6例雙側內聽道壺腹樣擴大,2例雙側內聽道狹窄;8例共10側頸靜脈球高位;8例蝶骨體發育不良;12例雙側外耳道向下傾斜,2例3側骨性外耳道閉鎖。眼眶:21例均表現為不同程度雙側眼眶外側壁夾角增大,眶距增寬,眼眶變淺,眼球突出,視神經迂曲。面中部:21例病例均有不同程度面中部凹陷,下頜骨相對前突;16例表現為鼻中隔偏曲;14例表現為腭弓高聳;2例表現為鼻淚管縮短,管徑增寬。頸椎:5例頸椎融合畸形;1例椎體蝴蝶椎。結論Crouzon綜合征顱CT表現紛繁多樣,診斷時多平面重建及三維影像結合觀察,可對這一少見疾病的正確診斷提供幫助。
Crouzon綜合征;體層攝影術;X線計算機
Crouzon綜合征是顱骨骨縫和頜面骨骨縫早閉引起的顱頜面部復合畸形的癥候群,由法國神經外科醫生Octave Crouzon于1912年首先報道。由于該病發病率低,除國內李建紅有8例CT影像總結報告外[1],國內外文獻多為個案報道,因此臨床醫生對該病多不熟悉。筆者收集分析21例近年來于上海第九人民醫院就診的Crouzon綜合征患者CT資料,以加強對該病的認識。
1 資料與方法
1.1 一般資料 收集2003-2010年于上海市第九人民醫院整形外科就診,臨床診斷為Crouzon綜合征的患者21例,其中男性13例,女性8例,年齡3~30歲,中位年齡18歲。
1.2 方法 所用CT機型為GE lightspeed16多層螺旋CT,層厚和層距均為1.25 mm,FOV 25 cm,掃描范圍從舌骨平面至顱頂。在Adw4.1工作站重組顱頜面冠狀面、矢狀面及三維影像,仔細觀察分析Crouzon綜合征患者顱頜面及頸椎異常CT表現。
2 結果
所有患者均表現為相似的CT特征,包括顱骨、眼眶、面中部等。
2.1 顱骨異常改變 5例舟狀頭畸形(圖1),4例尖頭畸形,3例短頭畸形;顱內壓增高癥候群:15例顱骨內板“指壓跡”,12例垂體窩增大(圖2);6例雙側內聽道壺腹樣擴大(圖3),2例雙側內聽道狹窄;8例共10側頸靜脈球高位;8例蝶骨體發育不良,雙側頸動脈管間距縮小(圖3);12例雙側外耳道向下傾斜(圖4),2例3側骨性外耳道閉鎖。
2.2 眼眶異常改變 21例均表現為不同程度雙側眼眶外側壁夾角增大,眶距增寬,眼眶變淺,眼球突出,視神經迂曲。筆者在Adw4.1工作站調整眼眶顯示平面至雙側眼球最大平面,雙側視神經基本全程顯示,分別測量眼球外側壁切線與中線的夾角(圖5),兩側相加得雙側眼眶外側壁夾角,21例病例最小98.9°,最大124.1°,平均109.3°。
2.3 面中部異常改變 21例病例均有不同程度面中部凹陷,下頜骨相對前突(圖2);16例表現為鼻中隔偏曲;14例表現為腭弓高聳;2例表現為鼻淚管縮短,管徑增寬,走向趨向水平(圖6)。

圖1 舟狀頭畸形,顱骨內板“指壓跡”,長箭示C2~C4融合,短箭示C5~C6融合;圖2 長箭示面中部凹陷,短箭示垂體窩增大,箭頭示C2~C4及C5~C6椎體多發融合;圖3 箭示雙側頸動脈管間距縮小,箭頭示雙側內聽道擴大
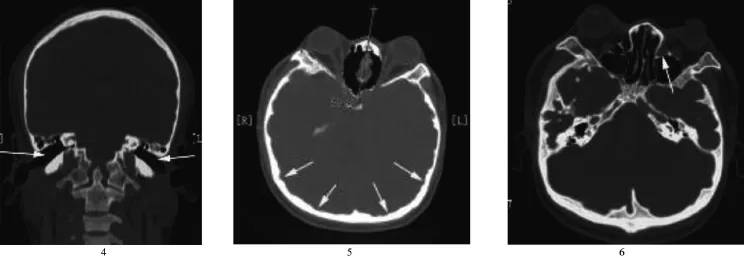
圖4 箭示雙側外耳道向下傾斜;圖5 雙側眼眶外側壁夾角增大,眶距增寬,眼眶變淺,眼球突出,箭示顱骨內板“指壓跡”;圖6 箭示鼻淚管管徑增寬
2.4 頸椎異常改變 5例頸椎融合畸形,其中3例C2~C3椎體融合,1例C2~C4及C5~C6椎體多發融合(圖2),1例C2~C3及C5~C6椎體多發融合,1例椎體蝴蝶椎。
3 討論
3.1 病因 Crouzon綜合征是一種罕見的由于顱縫早閉而引起的顱頜面部復合畸形的癥候群,是一種常染色體顯性遺傳病,全球出生嬰兒發病率約為1/25 000,是顱縫早閉癥中最常見的類型[2]。該綜合征是由第10號染色體的成纖維生長因子受體2 (Fibroblast growth factor receptor-2,FGFR-2)基因缺陷所致[3],發病患兒約半數有家族史,半數為基因突變所致。FGFR-2的過度表達可導致顱縫過早閉合,最常累及冠狀縫和矢狀縫。顱縫的過早閉合可影響顱骨、眼眶、上頜骨的發育。通常顱縫融合后,在其垂直方向上顱骨生長受限,而顱縫未閉合的顱骨可代償性生長以適應大腦的發育,從而導致異常的顱骨發育。多骨縫的早閉影響顱底骨的發育,導致眼眶及面中部發育的異常。
3.2 臨床表現 Crouzon綜合征患兒通常出生后就可發現顱頜面骨發育異常,并隨患兒生長發育而病情逐漸進展。臨床表現典型,如尖頭、舟狀頭、短頭畸形、突眼、眶距增寬、鷹鉤鼻、面中部凹陷、下頜骨相對前突等典型表現。此外患者可有傳導性耳聾、視力減退、斜視、打鼾等并發癥。當顱內高壓出現時,患兒可有頭痛、嘔吐、癲癇、腦積水、腦疝、智力發育障礙等一系列癥狀。
3.3 CT表現 Crouzon綜合征發生的根本原因是顱縫的過早閉合,不同顱縫的早閉會產生不同的頭顱形態改變。根據Virchow's法則,垂直于融合顱縫的顱骨生長方向受限,平行于融合顱縫的顱骨代償性生長[4]。當矢狀縫早閉,垂直于矢狀縫的側向生長受限,平行于矢狀縫的縱向生長代償增加,形成舟狀頭,本組病例中5例為舟狀頭畸形,表現為明顯向前突出的前額和向后突出的枕部,顱骨前后徑增加,左右徑減小,從側面觀察像一艘船的形狀。當雙側冠狀縫早閉,垂直于冠狀縫的縱向顱骨生長受限,側向生長代償性增加,形成短頭畸形。本組3例短頭畸形,表現為額骨凹陷,枕骨平直,顱骨前后徑減小,左右徑增大。當冠狀縫聯合矢狀縫或人字縫骨縫過早閉合,顱骨向上生長代償增加,形成尖頭畸形,本組4例尖頭畸形,表現為顱骨尖聳,前后及左右徑均減小。
當大腦生產速度快于顱骨生長的速度,患兒即可產生顱內高壓,CT表現為顱骨內板多發指壓跡改變,蝶鞍開口擴大,鞍底下陷,垂體窩擴大,垂體受壓縮小,嚴重者可有腦積水,表現為腦室顯著擴張。此外,李建紅等[1]在頭顱冠狀位觀察到視神經管可變扁變窄,本組也有2例符合上述改變。
當有多骨縫過早閉合時,顱底骨縫的早閉往往首先發生,可產生一系列的顱底、眼眶、面中部畸形改變。顱底骨的異常包括:蝶骨體發育不良,導致雙側頸動脈管聚攏;蝶骨大翼向前外側移位導致雙側眼眶外側壁夾角增大,眼眶變淺,這可能是由于由于顳側及顱底骨縫前后向生長不足所致[5]。筆者隨機選取10例年齡1~19歲(平均14歲)因外傷行頜面部CT檢查患者CT資料,測量雙側眼眶夾角,最小84.6°,最大96.0°,平均90.7°,明顯小于本組平均眼眶夾角109.3°
耳畸形在Crouzon綜合征患兒中較常見,臨床上可觀察到耳廓位置低,部分患兒并發傳導性耳聾。本組有12例表現為雙側外耳道向下傾斜,2例3側骨性外耳道閉鎖。胎兒耳的位置較成人低,在發育過程中耳逐漸上升到正常高度,Crouzon綜合征患者耳發育上升過程受阻,因此可導致外耳發育畸形。
頸椎的異常包括蝴蝶椎和頸椎椎體融合,頸椎融合的發生率約為18%,常累及C2~C3及C5~C6椎體[6]。本組3例C2~C3椎體融合,1例C5椎體蝴蝶椎,1例C2~C4、C4~C5椎體多發融合畸形,1例C2~C3、C5~C6椎體多發融合畸形,與文獻報道基本相符。Crouzon綜合征與Apert綜合征顱頜面表現相似,頸椎畸形的種類可作為一個重要的鑒別點。Hemmer發現Crouzon綜合征頸椎畸形常累及C2~C3椎體,而Apert綜合征C2~C3椎體多無融合,多累及C5~C6椎體[7]。
Crouzon綜合征顱頜面部CT表現紛繁多樣,診斷時多平面重建及三維影像結合觀察,可對這一少見疾病的正確診斷提供幫助。
[1]李建紅,王振常,鮮軍舫,等.Crouzon綜合征顱面部的CT表現(附8例報告)[J].臨床放射學雜志,2010,29(11):1461-1464.
[2]Ernest L,Bowling OD,Burstein FD.Crouzon syndrome[J].Optometry,2006,77:217-222.
[3]Rutland P,Pulleyn LJ,Reardon W,et al.Identical mutations in the FGFR2 gene cause both Pfeiffer and Crouzon syndrome phenotypes [J].Nat Genet,9:173-176,1995.
[4]Flores-Sarnat L.New insights into craniosynostosis[J].Semin Pediatr Neurol,2002,9(4):274-291.
[5]王世玉,呂長順.Crouzon綜合征的診斷及治療進展[J].中國美容醫學,2012,21(7):1273-1277.
[6]Anderson PJ,Hall CE,Evans RD,et al.Cervical spine anomalies in Crouzon syndrome[J].Spine,1997,22:402-405.
[7]Kaye M,Hemmer WH,Mcalister JL,et al.Cervical spine anomalies in the Craniosynostosis syndrome[J].Cleft Palate Journal,1987, 24:328-333.
3D CT analysis of 21 cases of Crouzon syndrome.
XIE Yu-zhi1,LIN Tao2,ZHU Ling1,ZHU Wen-jing1.
1. Department of Radiology,the Ninth People's Hospital of Shanghai Jiao Tong University School of Medicine,Shanghai 200011,CHINA;2.Shanghai Pudong New Area Zhoupu Hospital,Shanghai 201318,CHINA
ObjectiveTo discuss CT findings of Crouzon syndrome.MethodsThe imaging data of 3D skull CT in 21 cases with Crouzon syndrome were analyzed retrospectively.ResultsThe malformation involved skull,orbit,midface and cervical spine.The CT abnormalities of skull included scaphocephaly in 5 cases,oxycephaly in 4 cases,brachycephaly in 3 cases,diffuse indentation of inner table of skull in 15 cases,widening of hypophysial fossa in 12 cases,ampulla-type internal auditory canal in 6 cases,internal auditory canal stenosis in 2 cases,10 sides high riding jugular bulb in 8 cases,corpora ossis sphenoidalis dysplasia in 8 cases,downward sloping external acoustic meatus in 12 cases,3 sides atresia of external auditory canal in 2 cases.CT abnormalities of orbit included increased lateral orbital wall angle,hypertelorism,shallow orbit,proptosis and circuitous optic nerve.CT abnormalities of midface included concave midface and mandible relative to the protrusion in all 21 cases,septal deviation in 16 cases,hypoplasia,high-arched palate in 14 cases,shortening and widening naso-lacrymal duct in 2 cases.The CT abnormalities of cervical spine included cervical fusions in 5 cases,butterfly vertebrae in 1 case.ConclusionCT findings of Crouzon syndrome vary.Multiplanar reconstruction and 3D images combined can provide valuable information in diagnosing this rare disease.
Crouzon syndrome;Tomography;X-ray computed
R442.8
A
1003—6350(2015)02—0198—03
10.3969/j.issn.1003-6350.2015.02.0069
2014-06-19)
林 濤。E-mail:lintao9th@163.com
猜你喜歡
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
數學年刊A輯(中文版)(2022年4期)2022-02-16 08:17:34
今日農業(2021年19期)2022-01-12 06:16:36
中老年保健(2021年11期)2021-08-22 03:15:44
無線電通信技術(2021年4期)2021-07-13 08:58:28
無線電通信技術(2021年3期)2021-06-08 03:33:48
中學生數理化(高中版.高考數學)(2021年1期)2021-03-19 08:28:38
無線電工程(2020年11期)2020-10-29 01:25:46
現代出版(2020年3期)2020-06-20 07:10:34
福利中國(2015年4期)2015-01-03 08:03:38
