含氟雙二苯乙炔液晶的合成及液晶性
2014-08-21 09:04:56范程士羅忠林劉克剛聞建勛
化工生產與技術 2014年1期
范程士 羅忠林 劉克剛 聞建勛
(1.上海天問化學有限公司,上海200232;2.University Hospital Basel,Intensive Care Unit,Petersgraben,Basel44031)
為了滿足液晶平板顯示發展的各項要求,如響應速度快及器件視角寬的液晶材料,開發了具有不同特點顯示模式的顯示器。毫無疑問,具有正或者負的介電各向異性的向列型液晶材料是液晶顯示材料家族十分重要的成員。正性向列型液晶可用于標準的TFT-LCD(薄膜晶體管-液晶)顯示及IPS-LCD(面內開關-液晶)顯示(電場2極在同一塊板面上,施加電場使極性分子在電場中改變方向)。液晶分子軸方向與垂直分子軸方向的介電常數之差Δε為負數的稱為負性液晶,既可以在VA-TFT LCD(分子垂直排列的薄膜晶體管液晶顯示)模式中也可以在IPS模式中得到廣泛應用。
如果含氟向列型液晶具有高的雙折射性能,它們還可以在許多其他顯示技術方面得到重要應用。例如,膽甾相液晶具有電場為0時的多穩定相態織構現象,反射式膽甾液晶顯示,由于不需要偏振片及背光源,因此具有高反射能力及寬視角,特別適用于電子書籍及商業廣告等領域。還有在高技術的光子學應用領域,例如制造快速的快門、寬帶濾波器及全息攝影器件等。尤其是光子學領域的器件需要黏度低及雙折射大的混合液晶。這就是本工作的目的。
一般來說,沒有取代基的雙二苯乙炔有非常高的雙折射,但缺點是熔點高、對紫外線敏感、化學穩定性差。為此,本工作在結構上進行改造,在液晶核骨架上引入全氟亞苯基基團,對降低熔點、降低黏度有利,提高了化學穩定性,氟原子的范德華半徑與原子差別不大,保持非常寬的液晶溫度區域。側方向的2,3-二氟取代的導入,引入了2個強吸引電子的基團,結果形成優秀的負性向列型含氟液晶。可以分別以VA、IPS 2種不同工作模式應用于許多需要高速相應的光學器件中。優良的液晶原材料應具備高清亮點、液晶相范圍寬、高雙折射、黏度低、脂溶性好等特征,其中分子線性共軛性強的液晶化合物具有高雙折射率,例如多芳環類、炔類和多炔類化合物。
在上述化合物中,雙二苯乙炔類液晶的雙折射率n都在0.4以上,但是由于分子的共軛性很強造成此類化合物熔點很高,因此人們往往通過引入側基來降低熔點和黏度。2000年,C SHsu等人報道利用Sonogashira反應合成了含烷基邊鏈系列化合物,其分子特點是2端為烷基鏈,側向為短碳鏈烷基[1]。
氟原子的電負性強、體積小及脂溶性好等特點,在醫藥和材料上具有很廣泛的應用。筆者等在雙炔鍵的基礎上引入氟原子及四氟苯環特征基元以期降低熔點,增加向列相溫度范圍等特點設計、合成了一系列新穎含氟雙二苯乙炔類液晶,并對其相變性質進行研究。
結構通式:

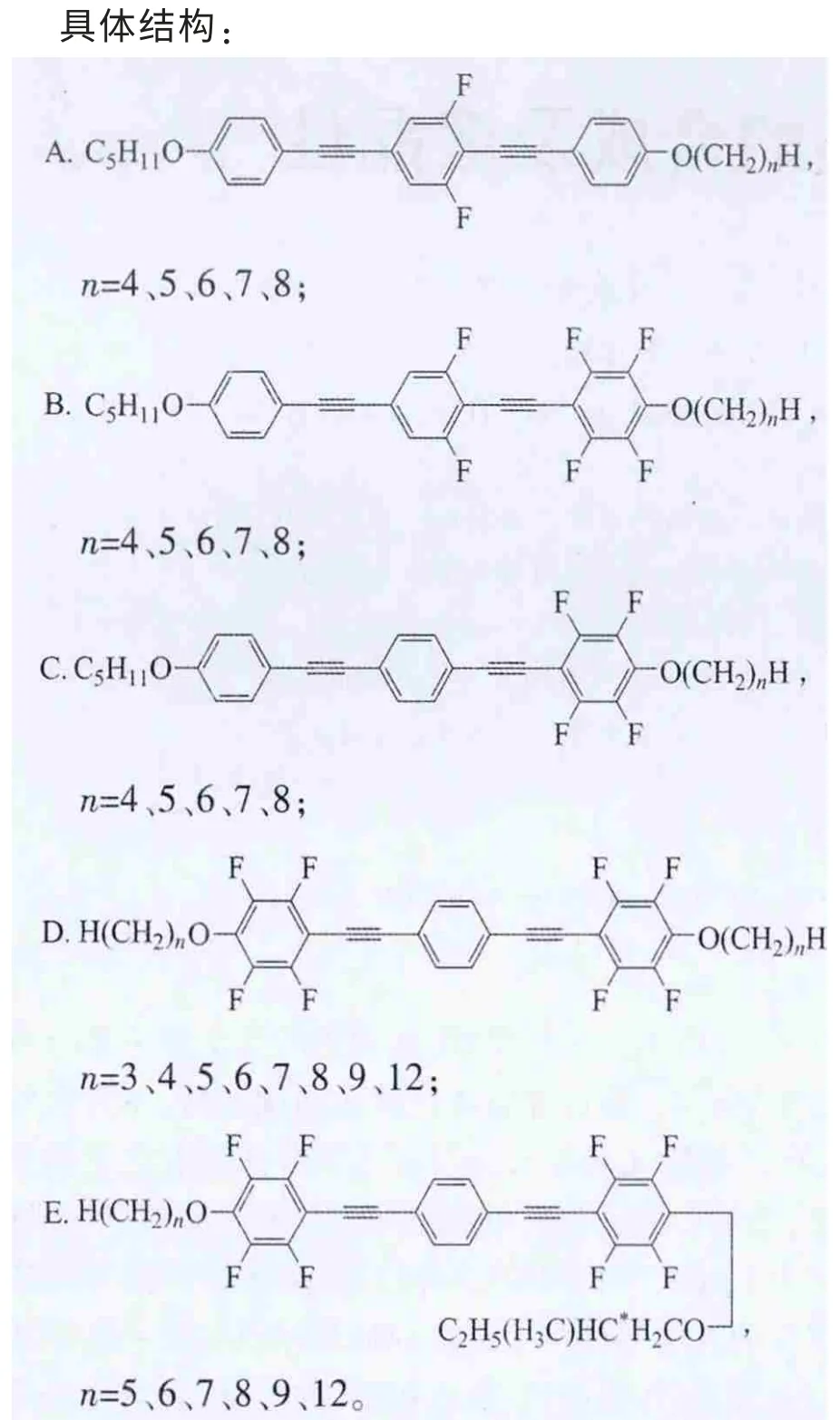
上述5類化合物的共同特點是:含有3個苯環,每2個相鄰的苯環之間以炔鍵連接,2端苯環末端以烷氧基鏈結束。其中A、B、C系列化合物的特點是苯環1的烷基R1鏈固定為正戊烷C5H11,另一端烷基R2鏈隨著碳原子數的變化具有不同長度[2-3]。它們的不同之處在于側邊苯環不同位置上引入氟原子:A系列是中間苯環2的2側對稱位置2個氫原子被2個氟原子取代;B系列是在A系列基礎上,靠近變化末端烷基的苯環3氫原子全部被氟原子取代;C系列是只有苯環3全氟代;D系列擁有對稱的結構,苯環1、3全氟代,2端具有相同的烷基鏈[4]。相對于D系列,E系列一端烷基鏈固定為手性戊基,而另一端為變化的正烷氧基鏈[5]。
1 化合物的合成路線
1.1 A、B、C系列
A、B、C系列化合物的合成路線:


合成此類化合物的關鍵步驟是在II價鈀和碘化亞銅催化下,炔鍵化合物和碘化物的偶聯反應。
化合物3的制備:以正戊氧基苯乙炔1為起始原料,在Pd(PPh3)2Cl2和CuI的催化下和3,5-二氟-1-碘苯偶聯得到中間體2,化合物2在正丁基鋰的作用下和碘反應得到關鍵中間體3。
化合物5的制備:同樣以正戊氧基苯乙炔1為起始原料,在Pd(PPh3)2Cl2和CuI的催化下和對碘苯胺偶聯得到中間體4,此化合物在亞硝酸鈉、濃鹽酸的氧化下得到偶氮化合物中間體,此中間體進一步和碘化鉀反應生成化合物5。
在Pd(PPh3)2Cl2和CuI的催化下,4-正烷氧基苯乙炔6和碘化物3偶聯得到目標化合物A;4-正烷氧基-2,3,5,6-四氟化合物7和碘化物3偶聯得到目標化合物B;4,-烷氧基-,3,5,6-四氟化合物7和碘化物5偶聯得到目標化合物C。
反應試劑和條件:a)3,5-二氟-1-碘苯,Pd(PPh3)2Cl2,CuI,Et3N;b)n-BuLi,I2,-78℃~室 溫;c)4-碘 苯胺,Pd(PPh3)2Cl2,CuI,Et3N;d)NaNO2,濃HCl,KI,四氫呋喃;e)Pd(PPh3)Cl2,CuI,Et3N。
1.2 D系列
D系列化合物的合成路線:

三甲基硅基五氟苯乙炔8在碳酸鉀的作用下和不同鏈長的正烷基伯醇發生反應得到中間體7,此炔基化合物在Pd(PPh3)2Cl2和CuI(碘化亞銅)的催化下和1,4-二碘苯發生雙偶聯反應得到目標化合物D。
反應試劑和條件:a)K2CO3,N,N-二甲基甲酰胺(DMF),室溫;b)Pd(PPh3)2Cl2,CuI,Et3N。
1.3 E系列
和上述提到的目標化合物的制備基本相似,E系列化合物的合成路線:
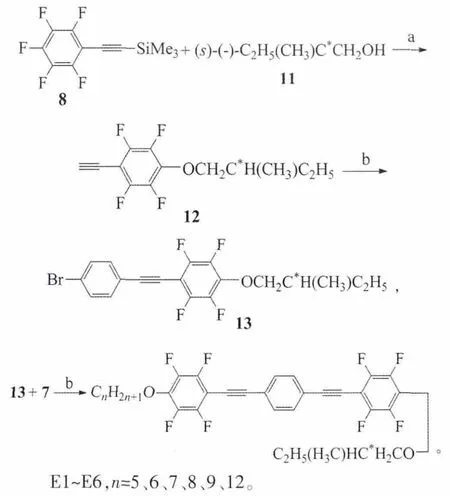
化合物8在碳酸鉀的作用下和手性醇11反應得到末端炔12,此化合物在Pd(PPh3)2Cl2和CuI的催化下和對溴碘苯發生偶聯反應生成溴化物13,同樣的條件下此溴化物和末端炔7偶聯提供目標分子E。
反應試劑和條件:a)K2CO3,DMF,室溫;b)Pd(Ph3)2Cl2,CuI,Et3N。
2 化合物的相變
通過偏光顯微觀察和差示掃描量熱法(DSC)對上述5個系列的液晶化合物的結構和性質進行系統分析。
2.1 A、B、C系列雙二苯乙炔類化合物
A、B、C系列化合物的相變溫度如表1所示,所有的化合物都呈現單一的向列相[2-3]。
A系列化合物擁有相同的結構主體,它們的不同之處在于一段烷氧基鏈的長度不同。總的來說,此類化合物顯示出很高的清亮點(從191~214℃);同時它們都含有很寬的向列相相變范圍(>97℃);另外,熔點和清亮點都隨著末端烷氧基鏈碳原子數的增加而逐漸降低。
B系列化合物含有1個四氟苯基官能團,此類化合物同樣顯示高的清亮點(從176~201℃);同時每一個化合物含有寬的相變范圍(>74℃);另外,熔點和清亮點都隨著末端烷氧基鏈碳原子數的增加而逐漸降低。
C系列化合物中間苯環沒有氟原子取代,此類化合物和A、B系列同樣擁有很高的清亮點(從184~211℃);同時每一個化合物含有很寬的相變范圍(>83℃);熔點和清亮點隨著末端烷氧基鏈碳原子數的增加先逐漸降低后增加。
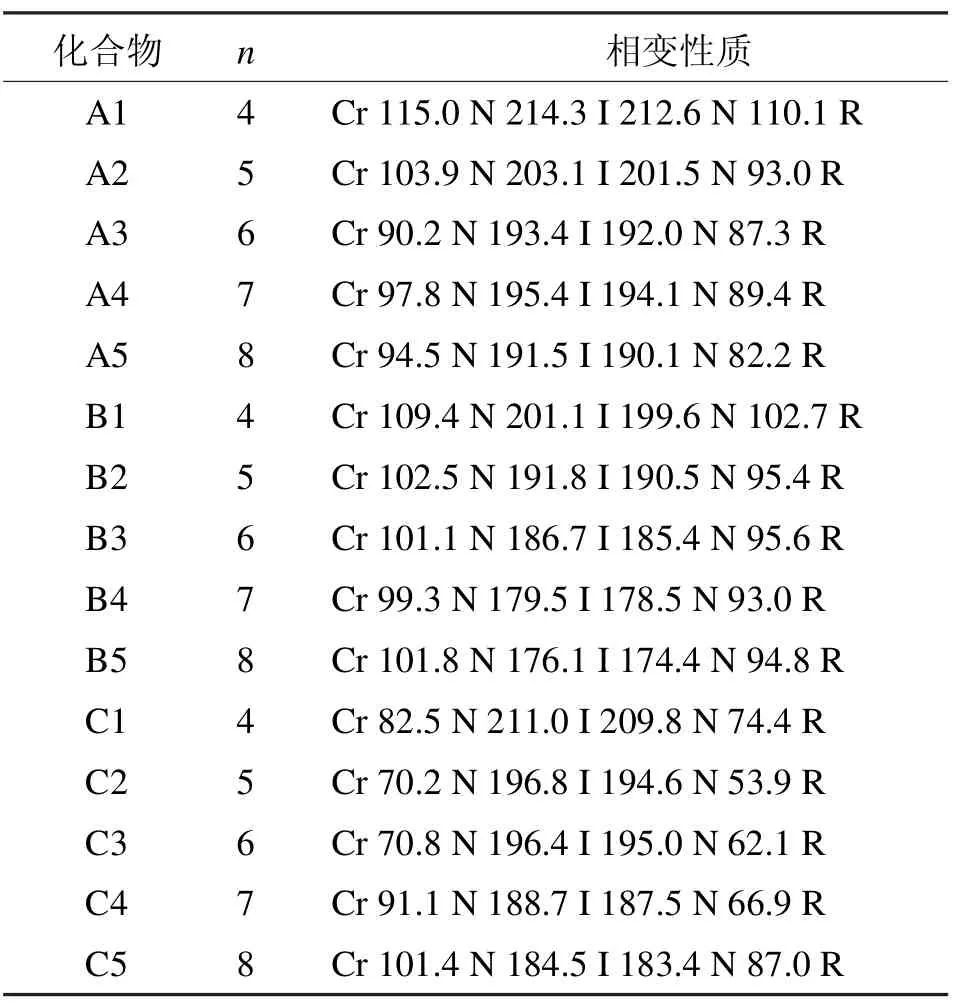
表1 A、B、C系列含氟雙二苯乙炔類液晶的相變性質Tab 1 Phase transition properties of liquid crystal of A,B,C series bisdiphenylacetylene which containing fluoride
通過A、B、C 3個系列化合物的相變數據分析可以看出,側面氟原子的取代對液晶的相變性質有很大的影響。在相同的末端烷氧基鏈的情況下,B系列化合物的熔點最低,而C系列的最高,清亮點則呈A>C>B趨勢。雙二苯乙炔類液晶共軛性很強從而擁有高的熱穩定性;當氟原子引入這個結構,因為氟原子的高電負性,它們之間或和氫原子之間的相互作用造成扭曲效應從而增加分子的厚度,熔點和清亮點從而降低。
2.2 D系列雙二苯乙炔類液晶
D系列化合物的相變性質如表2所示[4]。

表2 D系列含氟雙二苯乙炔類液晶的相變性質Tab 2 Phase transition properties of liquid crystal of D series bisdiphenylacetylene which containing fluoride
通過數據分析,8個化合物都顯示單一的向列相,碳鏈的長度和相變溫度呈現正常的奇偶效應,并且隨著烷氧基鏈的增加,熔點和清亮點都逐漸降低,另外,向列相的范圍先增加(烷氧基鏈碳原子個數n=3~6),隨后逐漸降低(烷氧基鏈碳原子個數n=7~12)。和已知文獻報道的苯環不含有氟原子并含有相同末端烷氧基鏈的雙二苯乙炔類液晶相比,D系列化合物熔點和清亮點降低,而且不支持近晶相(不穩定)[5]。
2.3 E系列雙二苯乙炔類液晶
E系列化合物的相變性質如表3所示[6]。
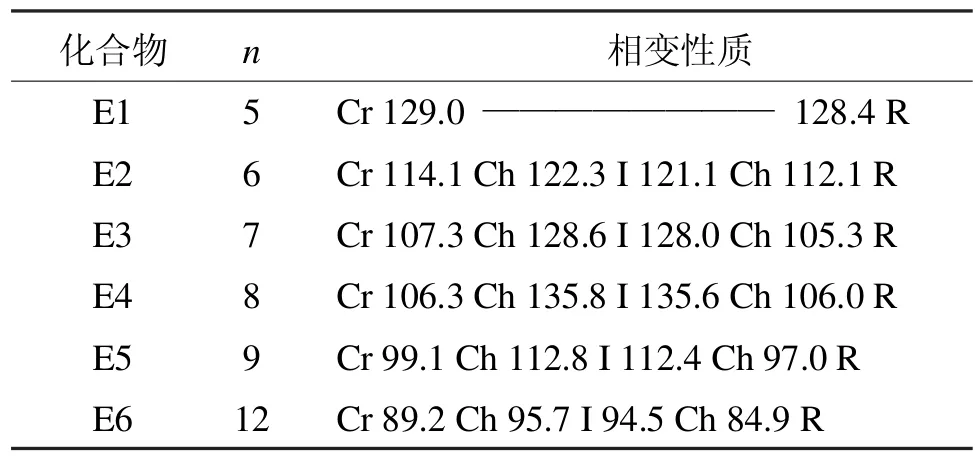
表3 E系列含氟雙二苯乙炔類液晶的相變性質Tab 3 Phase transition properties of liquid crystal of E series bisdiphenylacetylene which containing fluoride
除了烷氧基鏈為正戊烷氧基的化合物不呈現液晶態,其他化合物均具有單一的膽甾相。就末端烷氧基鏈的影響考慮,首先熔點隨著鏈長的增加而降低;而膽甾相變范圍是先增加(n=6-8)后降低。此類化合物的相變性質和D系列化合物相比,相變范圍變窄這說明支鏈化烷氧基鏈引起液晶的穩定性降低。
3 典型中間體和目標化合物的合成
3.1 4-正戊氧基-3′,5′-二氟二苯乙炔(2)
在一干燥的250 mL三頸瓶氮氣保護下加入4-正戊氧基苯乙炔5.0 g(26.6 mmol)、3,5-二氟-1-碘苯5.32 g(22.2 mmol)、Pd(PPh3)2Cl20.2 g(0.29 mmol)和CuI 0.1 g,隨后加入無水Et3N 100mL。反應混合液在30~40℃攪拌24 h。TLC跟蹤反應直至完全后,沉淀過濾乙醚萃取,有機相水洗,硫酸鎂干燥。真空除掉溶劑,硅膠柱層析得到白色固體7.23 g,產率91%。
1H NMR(CDCl3,TMS)(δ/ppm):7.56~7.18(m,7H),4.01(t,2H,J=5.4 Hz),1.90~1.34(m,6H),1.02(t,3H,J=3.7 Hz);19FNMR(CDCl3,TFA)(δ/ppm):32.6(m,2F)。
3.2 4-正戊氧基-3′,5′-二氟-4′-碘二苯乙炔(3)
4-正戊烷氧基-3′,5′-二氟二苯乙炔7.32 g(24.1mmol)溶解在84mL的THF中并冷卻到-78℃。向上述溶液中慢慢滴加13.3mL正丁基鋰(2 mol/L),此混合溶液在上述溫度下繼續攪拌1.5h。碘溶解在THF中,慢慢滴加到體系中,反應混合液升至室溫并繼續攪拌10h。隨后飽和氯化銨萃滅此反應。正常后處理得到粗產品,柱層析給出白色晶體5.8g,產率57%。
1HNMR(CDCl3,TMS)(δ/ppm):7.48~6.82(m,6H),3.98(t,2H,J=5.5Hz),2.01~1.27(m,6H),0.95(t,3H,J=3.6Hz);19FNMR(CDCl3,TFA)(δ/ppm):16.5(m,2F)。
3.3 4-正戊氧基-4′-氨基二苯乙炔(4)
合成路線和化合物4-正戊氧基-3′,5′-二氟二苯乙炔相似,得到化合物5.84g,產率79%。
1HNMR(CDCl3,TMS)(δ/ppm):7.42~6.61(m,8H),3.88(t,2H,J=5.5Hz),3,61(br,2H),1.79~1.27(m,6H),0.86(t,3H,J=3.7Hz)。
3.4 4-正戊氧基-4′-碘二苯乙炔(5)
4-正戊烷氧基-4′-氨基二苯乙炔2.8g(10.04 mmol)溶解在15mL的THF中并冷卻至0℃,濃鹽酸5.5mL和質量分數40%的亞硝酸鈉水溶液6.1 mL組成的混合液加入到上述溶液中,此體系在0℃下攪拌30min,然后加入6mol/L的KI水溶液17.3 mL,繼續攪拌3h,飽和硫代硫酸鈉20mL淬滅反應,正己烷萃取反應,正常的后處理,粗產品后處理得到白色晶體1.8g,產率50%。
1HNMR(CDCl3,TMS)(δ/ppm):7.42~7.72(m,8H),3.87(t,2H,J=5.4Hz),1.17~1.79(m,6H),0.86(t,3H,J=3.8Hz)。
3.5 1,3,-二氟-2-{2-[4-(正辛氧基)苯基]乙炔基}-5-{2-[4-(正戊氧基)苯基]乙炔基}苯(A5)
以化合物3和化合物6為偶聯原料,反應條件和過程如下面化合物的合成路線相似,得到目標化合物。
1HNMR(CDCl3,TMS)(δ/ppm):6.80~7.57(m,4H),3.99(t,4H,J=5.9Hz),0.85~1.99(m,24H);19FNMR(CDCl3,TFA)(δ/ppm):31.0(m,2F);MS(m/z):528(M+,100.0%),元素分析(EA):計算值:C79.52,H 7.24,F7.19;實際值:C79.60,H7.22,F7.18。
ABC系列其他化合物合成路線和此化合物相似。
3.6 1,4-二[(2,3,5,6-四氟-4-正辛烷苯基)乙炔基]苯(D6)
在一干燥50mL三頸瓶,氮氣保護下加入4-正辛氧基-2,3,5,6-四氟苯乙炔1.0g(3.3mmol)、對二碘苯0.55g(1.66mmol)、Pd(PPh3)2Cl230mg(0.043mmol)和CuI17mg(0.089mmol),隨后加入無水Et3N15mL。反應混合液加熱回流8h。TLC跟蹤反應直至完全后,沉淀過濾乙醚萃取,有機相水洗,硫酸鎂干燥。真空除掉溶劑,硅膠柱層析得到白色固體1.05g,產率95%。熔點120.0℃。
1HNMR(CDCl3,TMS)(δ/ppm):7.57(s,4H),4.30(t,4H,J=6.3Hz),0.74~2.00(m,30H);19FNMR(CDCl3,TFA)(δ/ppm):61.0(d,4F,J=18.6Hz),80.3(d,4F,J=18.6Hz);MS(m/z):678(M+,87.33%),454(100.00%),IR(KBr):2910、2840、1640、1520、1490、1440、1390、1140、1125、1020、1005、985、840、695、550cm-1;EA:C38H38F8O2,計算值:C67.26,H5.60,F22.42;實際值:C67.43,H5.72,F22.40。
D系列其他化合物合成路線和此化合物相似。
3.7 4-[(s)-2-甲基丁氧基]-2,3,5,6-四氟苯乙炔(12)
化合物三甲基硅基五氟苯乙炔6.0g(22.7 mmol)、碳酸鉀9.0g(65.1mmol)、(s)-2-甲基丁醇4.0g(45.5mmol)溶解在12mL的DMF中,此反應體系在40℃條件下攪拌46h然后60℃繼續攪拌6 h。常規后處理,粗產品柱層析得到產物為淺黃色液體5.40g(90.3%)。
1HNMR(CCl4,TMS)(δ/ppm):3.94(d,2H,J=6.0 Hz),3.34(s,1H),0.82~1.90(m,9H);19FNMR(CCl4,TFA)(δ/ppm):60.47(d,2F,J=18.8Hz),80.47(d,2F,J=18.8Hz)。
3.8 1-(4-溴苯基)-2-[4-((s)-2-甲基丁氧基)-2,3,5,6-四氟苯基]乙炔(13)
在一干燥100mL三頸瓶,氮氣保護下加入4-[(s)-2-甲基丁氧基]-2,3,5,6-四氟苯乙炔3.0g(11.8 mmol)、對溴碘苯3.34g(11.8mmol)、Pd(PPh3)2Cl2300mg(0.428mmol)和CuI163mg(0.857mmol),隨后加入60mL無水Et3N。反應混合液40℃下反應48h。TLC跟蹤反應直至完全后,沉淀過濾乙醚萃取,有機相水洗,硫酸鎂干燥。真空除掉溶劑,硅膠柱層析得到白色固體4.29g,產率88%。熔點45.5℃。
1HNMR(CCl4,TMS)(δ/ppm):7.40(s,4H),4.04(d,2H,J=6.0Hz),0.82~2.09(m,9H)。19FNMR(CCl4,TFA)(δ/ppm):60.00(d,2F,J=18.8Hz),79.50(d,2F,J=18.8Hz);MS(m/z):416(M+,43.6),414(M+,35.73),346(83.90),344(100.00)。
3.9 1-[(4-正戊氧基-2,3,5,6-四氟苯基)乙炔基]-4-[(4-((s)-2-甲基丁氧基)-2,3,5,6-四氟苯基)乙炔基]苯(E1)
在一干燥25mL三頸瓶,氮氣保護下加入1-(4-溴苯基)-2-[4-((s)-2-甲基丁氧基)-2,3,5,6-四氟苯基]乙炔374 mg(0.9 mmol)、4-正戊氧基-2,3,5,6-四氟苯乙炔235 mg(0.9 mmol)、Pd(PPh3)2Cl230 mg(0.043 mmol)和CuI 17 mg(0.089 mmol),隨后加入12mL無水Et3N。反應混合液回流8 h。TLC跟蹤反應直至完全后,沉淀過濾乙醚萃取,有機相水洗,硫酸鎂干燥。真空除掉溶劑,硅膠柱層析得到白色固體192 mg,產率56%。熔點129.0℃。
1H NMR(CCl4,TMS)(δ/ppm):7.45(s,4H),4.11(t,2H,J=5.0Hz),4.00(d,2H,J=5.0Hz),0.62~2.00(m,18H);19F NMR(CCl4,TFA)(δ/ppm):60.03(m,4F),79.75(m,4F);IR(KBr):2 960、2 870、2 200、1 520、1 505、1 490、1 440、1 390、1 130、985、840、690 cm-1;MS(m/z):594(M+,23.49),524(18.26),454(100.00)。
以下化合物的合成路線與此化合物相似。
3.10 1-[(4-正己氧基-2,3,5,6-四氟苯基)乙炔基]-4-[(4-((s)-2-甲基丁氧基)-2,3,5,6-四氟苯基)乙炔基]苯(E2)
產率56%。熔點114.1℃。
1H NMR(CCl4,TMS)(δ/ppm):7.46(s,4H),4.12(t,2H,J=5.0 Hz),4.01(d,2H,J=5.0 Hz),0.65~2.01(m,20H);19FNMR(CCl4,TFA)(δ/ppm):60.05(m,4F),79.77(m,4F);IR(KBr):2 960、2 870、2 200、1 520、1 505、1 485、1 438、1 390、1 125、982、840、688 cm-1;MS(m/z):608(M+,66.74),538(25.79),454(100.00)。
3.11 1-[(4-正庚氧基-2,3,5,6-四氟苯基)乙炔基]-4-[(4-((s)-2-甲基丁氧基)-2,3,5,6-四氟苯基)乙炔基]苯(E3)
產率58%。熔點107.3℃。
1H NMR(CCl4,TMS)(δ/ppm):7.48(s,4H),4.12(t,2H,J=5.0 Hz),4.01(d,2H,J=5.0 Hz),0.67~2.01(m,22H);19FNMR(CCl4,TFA)(δ/ppm):60.04(m,4F),79.76(m,4F);IR(KBr):2 960、2 870、2 200、1 520、1 505、1 490、1 440、1 390、1 128、982、840、690 cm-1;MS(m/z):622(M+,77.90),552(29.03),454(100.00)。
3.12 1-[(4-正辛氧基-2,3,5,6-四氟苯基)乙炔基]-4-[(4-((s)-2-甲基丁氧基)-2,3,5,6-四氟苯基)乙炔基]苯(E4)
產率49%。熔點106.3℃。
1H NMR(CCl4,TMS)(δ/ppm):7.50(s,4H),4.14(t,2H,J=5.0 Hz),4.00(d,2H,J=5.0 Hz),0.66~2.00(m,24H);19F NMR(CCl4,TFA)(δ/ppm):60.03(m,4F),79.75(m,4F);IR(KBr):2960、2870、2200、1 520、1 505、1 495、1 441、1 395、1 130、990、840、694 cm-1;MS(m/z):636(M+,24.33),566(11.21),454(100.00)。
3.13 1-[(4-正壬氧基-2,3,5,6-四氟苯基)乙炔基]-4-[(4-((s)-2-甲基丁氧基)-2,3,5,6-四氟苯基)乙炔基]苯(E5)
產率49%。熔點99.1℃。
1H NMR(CCl4,TMS)(δ/ppm):7.48(s,4H),4.12(t,2H,J=5.0 Hz),4.00(d,2H,J=5.0 Hz),0.68~2.00(m,26H);19FNMR(CCl4,TFA)(δ/ppm):60.04(m,4F),79.77(m,4F);IR(KBr)(cm-1)2 960、2 870、2 200、1 520、1 505、1 495、1 440、1 394、1 130、990、840、694;MS(m/z):650(M+,25.11),580(15.17),454(100.00)。
3.14 1-[(4-正十二氧基-2,3,5,6-四氟苯基)乙炔基]-4-[(4-((s)-2-甲基丁氧基)-2,3,5,6-四氟苯基)乙炔基]苯(E6)
產率56%。熔點:89.2℃。
1H NMR(CCl4,TMS)(δ/ppm):7.49(s,4H),4.10(t,2H,J=5.0 Hz),4.00(d,2H,J=5.0 Hz),0.70~2.00(m,32H);19F NMR(CCl4,TFA)(δ/ppm):60.06(m,4F),79.80(m,4F);IR(KBr):2 960、2 870、2 200、1 520、1 505、1 495、1 440、1 394、1 130、990、840、694cm-1;MS(m/z):692(M+,49.32),622(24.20),454(100.00)。
4 結論
在雙苯二乙炔的骨架引入氟原子及四氟亞苯基基元,設計合成了A~E 5個系列的含氟雙二苯乙炔液晶,利用Sonogashira交叉偶合反應完成了適于工業化的合成方法。用附帶加熱臺的偏光顯微鏡及DSC方法,測定了它們的液晶性。
研究發現:1)側向氟原子取代可以抑制近晶相的出現(A);2)單一的四氟亞苯基有利于抑制近晶相出現,形成溫度200℃以上、高清亮點、低熔點液晶化合物,盡管與不含氟母體化合物比較,清亮點有所下降,但是在應用方面是可以接受的(C);3)在化合物C增加對稱的2個氟原子,形成的B化合物,液晶性下降,原因是分子間引力進一步減弱;4)在化合物D中有2個四氟亞苯基,分子間引力與化合物C比較,大大減弱,不僅清亮點下降,而且液晶溫度區間大大變窄;5)側鍵為手性基團的化合物E(見表3),與它的母體化合物D比較,其液晶性幾乎遭到破壞。
應用Sonogashira交叉偶合法建立的合成方法,不僅可以合成結構對稱的分子,也可以合成結構不對稱的分子。可以工業化生產。
化合物A與B是2類介電常數正極性液晶,化合物C是介電常數非極性液晶,它們有高清亮點及足夠寬的溫度區間。氟取代基的強烈拉電荷作用,使它們避免了非含氟化合物母體的化學不穩定,提高了抗氧化性。由于保留了母體的大的光學雙折射性能,進一步降低黏度,并且與上述各種優點結合,有利于制備高性能的液晶混合物,可以用于光子學方面的液晶顯示技術。
[1]C SHsu,K F Shyn,Y Y Chang,et al.Synthesis of laterally substituted bistolane liquid crystals[J].Liquid Crystals,2000,27:283-287.
[2]聞建勛,劉克剛,李衡峰.含氟雙二苯乙炔類化合物、制備方法及用途:中國,1293180[P].2001-05-02.
[3]Kegang Liu,Hengfeng Li,Kan Wang,et al.Synthesis and characterization of novel fluorinated bistolane-type liquid crystals[J].Liquid Crystals,2001,28:1463-1467.
[4]Yuelian Xu,Yueqing Hu,Qi Chen,et al.Synthesis and characterization of octafluorinated 1,2-(4,4′-dialoxyaryl)acetylene monomers and 1,4-bis[4′,4″-dialkoxyphenyl)ethynyl]benezene dimmers[J].JMater Chem,1995,5:219-221.
[5]CPugh,SK Andersson,V Percec.Phase transfer Pd(0)/Cu(I)catalysedpolymerizationreactions7.Synthesisand thermotropic behaviourof1,4-bis[2-(3′,3″-difluoro-4′,4″-di-n-alkyloxyphenyl)-ethynyl]benzene dimers[J].Liquid Crystals,1991,10:229-232.
[6]JianxunWen,Minquan Tian,QiChen.Novel fluorinated liquid crystals.Part VI.The synthesis and phase transition of novel cholesteric liquid crystals containing 1,4-tetrafluorophenylene units[J].Journal of Fluorine Chemistry,1994,68:117-120.
