PI3K/Akt/GSK3β信號通路及內質網應激蛋白在大鼠難免性壓瘡形成中表達變化
2014-08-08 09:41:12崔飛飛張菊芳吳海英宋亞軍蔣美琴翁玉英姜麗萍
溫州醫科大學學報 2014年8期
崔飛飛,張菊芳,吳海英,宋亞軍,蔣美琴,翁玉英,姜麗萍
壓瘡(又稱壓迫性潰瘍)分淺表性潰瘍和深層潰瘍,2007年美國壓瘡顧問小組(National Pressure Ulcer Advisory Panel,NPUAP)將深層潰瘍重新定義為深部肌肉組織損傷,臨床上稱其為“惡性壓瘡”或“難免性壓瘡”[1-2]。研究發現,肌肉損傷后其恢復和細胞凋亡是同步進行的,倘若細胞凋亡持續存在,則不利于肌肉的恢復[3]。因此,肌肉細胞凋亡在難免性壓瘡恢復進程中發揮重要潛在作用。研究發現,內質網介導的細胞損傷在凋亡過程中發揮著重要作用,其中磷脂酰肌醇3-激酶(phosphatidylinositol-3-kinases,PI3K)/蛋白激酶B(protein serine threonine kinase,Akt)/糖原合成激酶3β(glycogen syn-thase kinase3β,GSK3β)信號通路是維持細胞正常生存和功能的重要信號轉導途徑,在缺血性損傷和再灌注損傷中起明顯的保護作用[4]。目前在腫瘤、心、腦缺血-再灌注損傷等領域研究較多,但PI3K/Akt/GSK3β信號通路在難免性壓瘡中的研究鮮見報道。本研究通過動物模型來觀察、探討PI3K/Akt/GSK3β信號通路和內質網分子伴侶在難免性壓瘡形成過程中的表達變化及作用。
1 材料和方法
1.1 動物及主要試劑 健康成年雄性SD大鼠54只(上海斯萊克實驗動物有限責任公司提供),體質量320~350 g。大鼠自由飲水、進食,室溫(25±2)℃,分籠喂養。抗體葡萄糖調節蛋白78(GRP78)、轉錄因子C/EBP同源蛋白(CHOP)、Akt、P-Akt、GSK3β、P-GSK3β由Santa Cruz Biotechnology提供,天冬氨酸特異性半胱氨酸蛋白酶12(caspase-12)、半胱氨酸天冬氨酸蛋白酶-3(caspase-3)抗體從Abcam公司購買,二抗和內參抗體分別由上海碧云天生物技術研究所和武漢博士德生物工程有限公司提供。
1.2 動物模型制備
1.2.1 難免性壓瘡(可疑深部組織損傷)判斷標準[5]:2007年,NPUAP對壓瘡深部肌肉組織損傷的診斷標準:皮下軟組織受到壓力或剪切力的損害,局部皮膚完整但可出現顏色改變如紫色或褐紅色,或導致充血的水皰。與周圍組織比較,這些受損區域的軟組織可能有疼痛、硬塊、滲出、潮濕、發熱或冰冷(見圖1)。
1.2.2 模型制備及分組:參照王艷艷等[6]的方法制備壓瘡深部組織損傷動物模型,來模擬臨床難免性壓瘡發生的背景。用10%水合氯醛按300 mg/kg腹腔注射麻醉后,將大鼠俯臥于泡沫床墊上,用剪毛刀對其大腿兩側股薄肌部位進行剪毛,暴露施壓部位。按照隨機數字表法將大鼠分為正常對照組(Con組),1個周期受壓組(1c組)、3個周期受壓組(3c組)、6個周期受壓組(6c組)、9個周期受壓組(9c組)和恢復期1 d、3 d、5 d、7 d組,每組各6只。各組大鼠體質量與Con組相比,差異無統計學意義。本實驗模擬臨床壓瘡發生的處理為于大鼠兩側大腿股薄肌施加22.47 kPa壓力,持續施壓2 h,放松0.5 h,即1c組(見圖1C-D)。1c組大鼠模擬壓瘡處理1次;3c組大鼠1 d內模擬壓瘡處理3次,只處理1 d;6c組大鼠1 d內模擬壓瘡處理3次,連續處理2 d;9c組大鼠1 d內模擬壓瘡處理3次,連續處理3 d;恢復期組為大鼠9c組后,解除壓力,分別于第1、第3、第5、第7天,觀察局部受壓組織的變化,Con組大鼠未行施壓處理。各組大鼠于施壓處理結束后給予安樂處死。
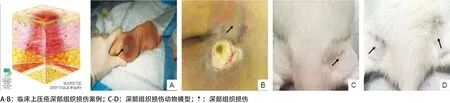
圖1 臨床和動物深部組織損傷模型及案例
1.3 檢測方法 在實驗終點,在冰上迅速切取各組受壓中心部位肌肉組織,于4 ℃ 0.9%氯化鈉溶液中快速洗去血液,用濾紙吸干水分并且剪碎后于 -80 ℃超低溫冰箱保存備用。另取相同部位受壓肌肉組織(0.5 cm×0.5 cm×0.5 cm),4 ℃ 0.9%氯化鈉溶液洗凈血液后,4%的多聚甲醛-PBS液固定,脫水,常規石蠟包埋,蠟塊連續切片5μm厚,進行組織形態學分析。凍存的所有樣品于研究結束時,進行勻漿,離心(4 ℃,12 000 r/min,15 min)收集上清液用于蛋白質免疫印跡(Western blotting)法 檢測。
1.3.1 組織形態學觀察:組織常規脫蠟,梯度透明,HE染色,用Nikon(TE 2000-U)顯微鏡觀察,NIS Elements圖像采集軟件采集圖像,觀察受壓組和恢復組在不同階段局部受壓肌肉組織病理學變化。
1.3.2 免疫組織化學法:采用SABC法,常規設立陰性對照。3% H2O2處理后熱修復抗原,5% BSA封閉,一抗(1:400)4 ℃過夜,生物素化二抗37 ℃孵育30 min,DAB顯色,蘇木精復染,中性樹膠封片,鏡下進行定性觀察。每張組織切片在×20物鏡下觀察4個視野(不重復),計數陽性細胞核數,以每平方毫米的陽性細胞核數(n/mm2)來表示。
1.3.3 Western blotting法:取各組大鼠肌肉組織各200 mg(于冰上進行操作),加入0.5 mL組織蛋白裂解液ZD408,電動勻漿器將組織研碎,4 ℃離心,取上清,按Bradford法測量蛋白濃度。每孔上樣20μL,進行十二烷基硫酸鈉-聚丙烯酰胺凝膠電泳,恒壓轉膜,30 V恒壓預跑15 min,80 V恒壓電泳跑至濃縮膠和分離膠分界線,120 V恒壓電泳90 min。再120 V恒壓轉膜90~120 min。50 g/L脫脂奶粉封閉1 h,加入一抗(GRP78、CHOP、Akt、p-Akt、GSK3β、p-GSK3β的一抗濃度為1:300,caspase-12和 GAPDH的一抗濃度為1:1 000)孵育,4 ℃過夜。Tris-吐溫20緩沖液洗滌,加入辣根過氧化物酶標記二抗(GRP78、CHOP的二抗濃度為1:1 000,caspase-12、Akt、p-Akt、GSK3β、p-GSK3β和GAPDH的二抗濃度為1:2 000),室溫孵育1 h,洗滌后,暗室中加化學發光顯色液顯色,X線膠片曝光,常規方法顯影、定影。以GAPDH作為內參,采用Quantity One灰度分析軟件分析條帶的灰度值,結果以目的蛋白GRP78、CHOP、caspase-12與GAPDH內參比值表示,p-Akt、p-GSK3β蛋白含量用p-Akt/Akt,p-GSK3β/GSK3β表示。本實驗重復進行3次。
1.4 統計學處理方法 采用SPSS 16.0統計軟件進行單因素方差分析,數據以±s表示,兩兩比較采用LSD-t法。P<0.05為差異有統計學意義。
2 結果
2.1 HE染色觀察各組肌肉形態學變化 光鏡下HE染色觀察受壓肌肉組織顯示:Con組肌纖維排列緊密,形態正常,結構清晰,骨骼肌橫紋清晰,無明顯的炎性細胞浸潤(見圖2A),與Con組相比,隨著受壓周期的延長,受壓組肌肉組織逐步出現退化的病理變化。表現為肌纖維排列紊亂、水腫,肌間隙增寬并出現少量的碎片(見圖2B-C),間質區域出現炎癥細胞浸潤逐漸加重,肌纖維出現溶解、斷裂,壞死碎片,空泡變性,玻璃樣變性等病理改變(見圖2C-E);恢復期1 d組發現在肌纖維溶解、斷裂,壞死區域有大量的炎性細胞聚集(見圖2F),隨后的炎性細胞逐漸減少,但還存在大量的肌纖維紊亂、斷裂,壞死碎片等病理變化(見圖2G-I)。
2.2 受壓肌肉組織中caspase-3蛋白在不同階段的 表達 免疫組織化學檢測結果顯示:caspase-3免疫 陽性產物呈棕黃色顆粒,主要位于胞漿。caspase-3蛋白在Con組中呈弱表達,隨著時間延長,其蛋白表達逐漸增加,9c組表達顯著;恢復期1 d組表達達高峰,隨后表達開始下降,見圖3。
2.3 Western blotting法檢測內質網應激相關蛋白的表達 檢測結果顯示:與Con組相比,蛋白GRP78、CHOP、caspase-12的表達表現為損傷后呈上升趨勢,GRP78在3c組出現第一次峰值(P<0.05),恢復期1 d組表達顯著(P<0.05),5 d組出現第二次峰值(P<0.01),7 d組開始下降(P<0.05),但仍高于Con組;CHOP在1c組表達顯著(P<0.05),9c組出現第一次高峰(P<0.01),恢復期1 d、3 d組表達下降(P<0.05),5 d組表達又開始上升(P<0.05),7 d組表達達高峰(P<0.01);caspase-12在6c組表達顯著(P<0.05),9c組出現第一次峰值(P<0.01),1 d、3 d組持續高表達(P<0.05),之后略有下降(P<0.05),但仍明顯高于Con組,見圖4。

圖2 不同組受壓肌肉組織的組織改變(HE,×200)
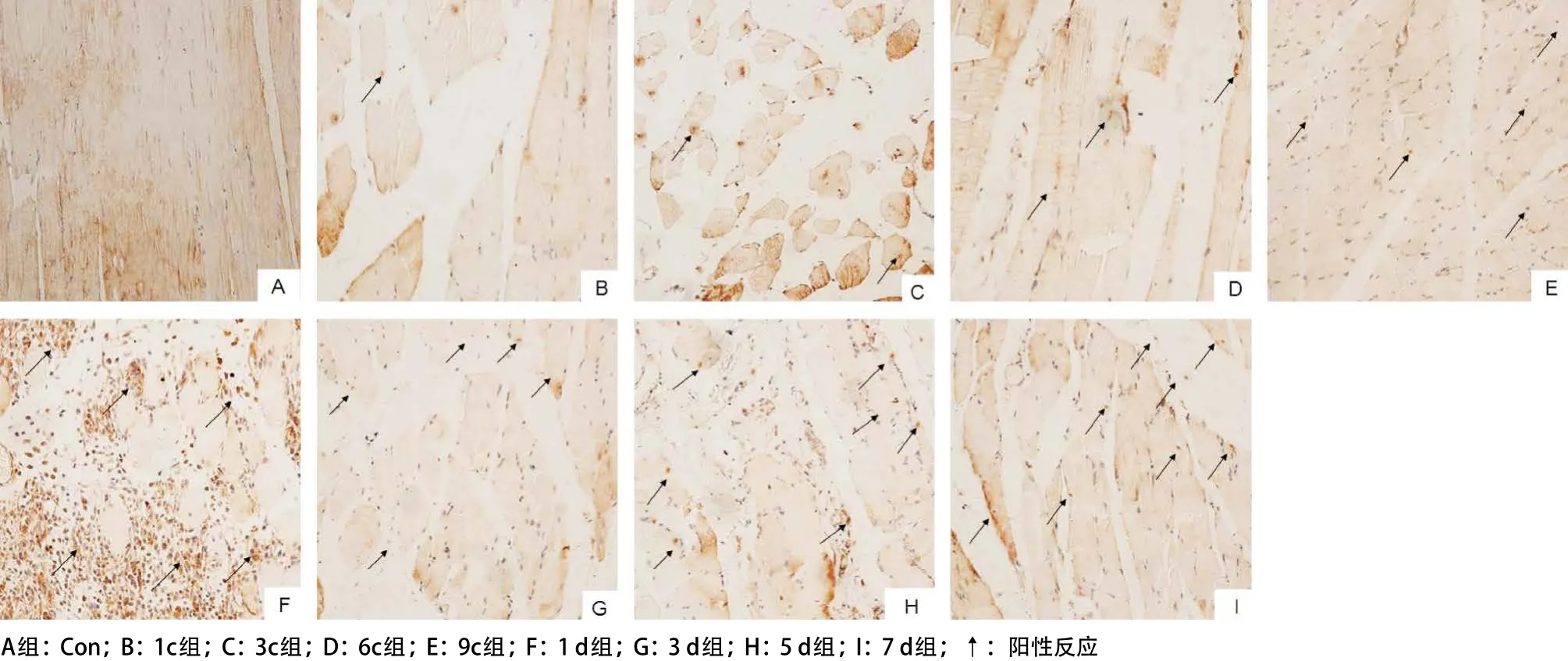
圖3 不同組受壓肌肉組織中的caspase-3的表達(IHC,×200)
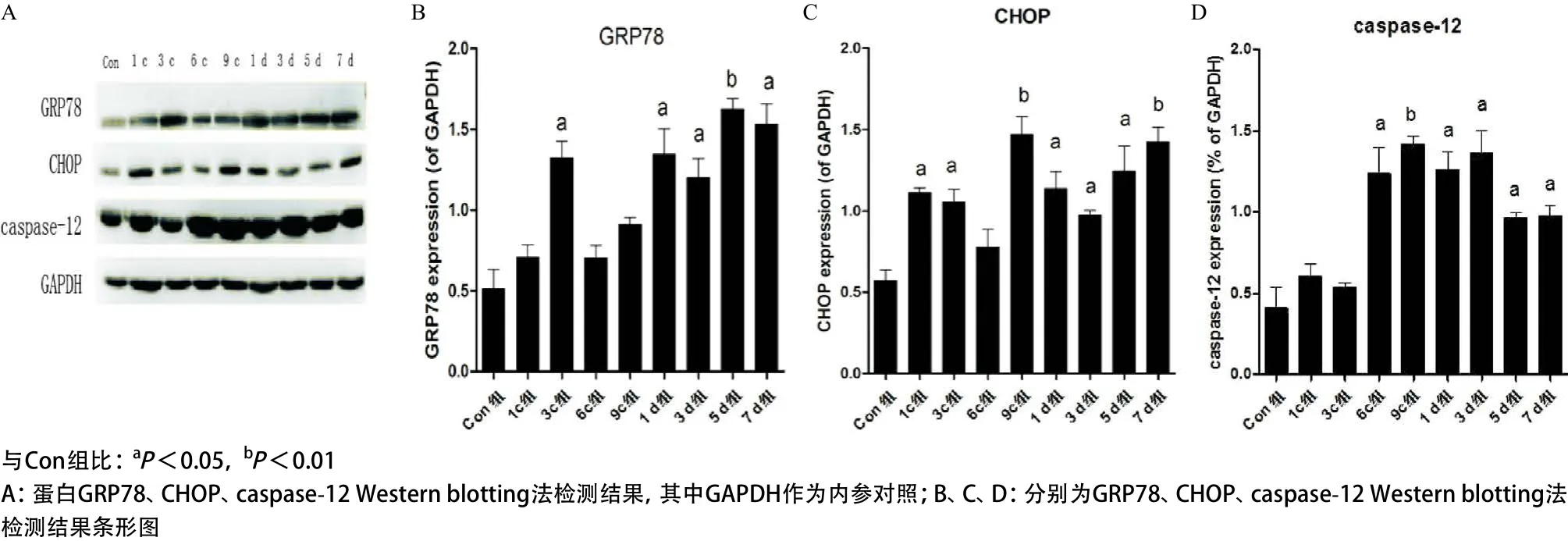
圖4 不同組受壓肌肉組織中GRP78、CHOP、caspase-12的表達
2.4 Western blotting法檢測Akt、GSK3β蛋白及其磷酸化程度 檢測結果顯示:與Con組相比,受壓組和恢復組p-Akt蛋白表達呈先上升后下降的趨勢。1c組,p-Akt表達顯著(P<0.01),9c組蛋白表達下降最為明顯(P<0.001);1 d組表達開始增加(P<0.001),3 d組表達呈現第一次高峰(P<0.001);p-GSK3β蛋白表達呈先上升后下降的趨勢,在1c、3c組,表達開始升高(P<0.05),6c組表達開始下降,但差異無統計學意義(P>0.05),9c組蛋白表達下降最為明顯(P<0.05);恢復組p-GSK3β蛋白表達呈先下降后上升趨勢,表現為與9c組相比,1 d組略有下降(P<0.05),3 d組下降明顯(P<0.01),5 d 組表達開始增加(P<0.01),7 d組表達出現高峰,見圖5。
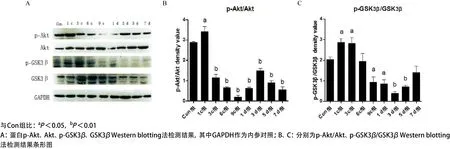
圖5 不同組受壓肌肉組織中總的Akt、GSK3β及其磷酸化的程度
3 討論
3.1 探索難免性壓瘡損傷的細胞信號通路,可為臨床預防和治療提供干預靶點 壓瘡的發生及發展的細胞分子機制仍不明確,給臨床預防和治療壓瘡帶來了很大的困難。因此,對壓瘡損傷的細胞信號通路進行研究,尋找治療的分子靶點,可為臨床防治壓瘡提供新思路、新方法。研究表明,細胞凋亡機制成為組織損傷和修復領域的研究熱點之一,尤其在皮膚創傷與愈合,慢性潰瘍形成與愈合的研究領域得到了大量的驗證。研究發現,ERS誘發的凋亡是一種細胞凋亡新途徑。本研究通過觀察難免性壓瘡形成和修復過程中細胞凋亡因子caspase-3、GRP78、CHOP、caspase-12及其Akt、GSK-3β磷酸化程度,觀察PI3K/Akt信號通路及其下游分子GSK3β在難免性壓瘡損傷中的表達變化,探討細胞凋亡在難免性壓瘡形成過程中的潛在作用及其細胞發生凋亡的信號通路,以尋找干預壓瘡的分子靶點。
HE染色結果表明,隨著受壓周期的延長,由于缺血性損傷作用使受壓肌肉組織出現逐步退化病理變化。具體表現為,受壓肌肉組織炎癥細胞增多,肌間隙增寬,肌纖維出現水腫、溶解、斷裂、空泡化、玻璃樣改變現象,在恢復組,由于缺血、低氧條件的改善,受壓組織炎性細胞浸潤程度逐漸減輕,但肌纖維恢復速度較慢,仍然存在肌纖維斷裂、壞死片段等病理現象,與臧爽等[7]研究結果一致。
3.2 內質網分子伴侶蛋白在難免性壓瘡中的表達及作用 ERS介導的細胞存亡具有環境刺激依賴性,應激的強度和持續時間決定了細胞是否能夠恢復和維持內質網的穩態。適宜的ERS可誘導GRPs、鈣網蛋白(calreticulin,CRT)、蛋白質折疊酶等物質的表達上調,增強細胞耐受應激刺激能力[8],而持續嚴重的ERS則誘導CHOP、caspase-12等促凋亡因子的表達及活化,觸發內質網凋亡信號途徑,導致細胞凋亡和組織損傷[9]。
本研究實驗組GRP78、CHOP、caspase-12的表達與前期研究結果[10-11]趨勢一致,但其在恢復期變化情況卻不清楚。本實驗發現,免疫組織化學法檢測caspase-3表達呈上升趨勢,具體表現為9c組出現第一次高峰,恢復期1 d、3 d組出現持續高表達,隨后出現下降趨勢;恢復組GRP78、CHOP、caspase-12 表達整體上呈上升趨勢,具體表現為1 d組GRP78表達顯著,CHOP蛋白表達下降,筆者認為,可能與損傷后恢復期間,由于損傷后早期的再灌注損傷,產生大量的自由基,使機體處于氧化應激狀態,從而誘導ERS持續存在,起細胞保護作用的GRP78高表達可以處理機體的錯誤折疊蛋白或未折疊蛋白,使ER內穩態恢復平衡;5 d組GRP78出現第二次峰值, 7 d組出現下降趨勢,而CHOP蛋白在5 d組表達下降,7 d組表達出現第二次峰值,筆者認為可能與自由基的連鎖反應使機體持續處于氧化應激狀態,導致ER腔積聚過多的錯誤折疊蛋白,超過機體的處理能力,激活下游CHOP蛋白表達,處理過度受損的細胞,這與左群等[12]研究結果趨勢一致;而恢復組caspase-12在1 d、3 d組持續高表達,之后出現下降趨勢,前期出現持續高表達,可能與其特有的通路有關,后期出現下降趨勢,這與免疫組織化學caspase-3的表達趨勢一致。
3.3 PI3K/Akt/GSK3β信號通路對細胞的調節作用是否與難免性壓瘡損傷存在聯系 近年來,越來越多的研究[3]報道,肌肉損傷恢復情況與肌細胞凋亡存在密切的關系,但具體的細胞凋亡信號通路仍不清楚。研究表明,PI3K/Akt、GSK-3β通路是調節細胞增殖、分化、凋亡和衰老的關鍵途徑[4]。
Akt是一種絲氨酸/蘇氨酸激酶,它是PI3K下游最重要的效應遞質,能調節各種細胞內的信號通路。活化的Akt磷酸化后,進一步激活或抑制其下游的靶蛋白,進而發揮其調節細胞增殖、分化,葡萄糖代謝以及遷移等作用。當組織細胞遭受缺血-再灌注損傷時,活化的Akt還可以防止細胞凋亡,促進細胞的存活[13]。
GSK3β是Akt下游的一種活性激酶,主要是通過抑制其活性來調節其功能的。GSK3β不僅能調節糖代謝,而且在細胞增殖、生長及死亡中也發揮了重要作用。當組織面臨缺血性損傷和再灌注損傷時,GSK3β活化會促進caspase-3的激活和細胞色素c的釋放,促進細胞程序性死亡,加重損傷。
本研究Western blotting法檢測發現,隨著受壓周期次數的延長,p-Akt蛋白表達呈逐漸下降的趨勢,p-GSK3β蛋白表達呈先上升后下降的趨勢,恢復組p-Akt蛋白表達呈上升趨勢,p-GSK3β蛋白表達呈先下降后上升趨勢。筆者推測,由于外面施加的機械性壓力,激活了PI3/Akt信號通路,致使1c組,p-Akt表達出現第一次高峰,隨著受壓周期次數的增加,缺血再灌注損傷加重,抑制了Akt活性,使Akt磷酸化程度下降;9c組Akt磷酸化程度下降最為明顯,細胞凋亡最為明顯,與CHOP、caspase-12、caspase-3的表達成反比;Akt活性被抑制,從而抑制了GSK3β的ser9位點進行磷酸化,表現為p-GSK3β呈下降的趨勢,促進了MPTP開放,促進細胞凋亡;恢復組由于解除了外部機械性壓力和缺血性損傷的改善,逐漸恢復了Akt的活性,但仍然不能明顯改善細胞凋亡的命運,這與Mozaffari等[14]的研究結果一致。
總之,難免性壓瘡之所以難以恢復可能與PI3K/Akt/GSK3β信號通路和內質網應激介導的細胞凋亡有關,這將為我們接下來的干預實驗提供理論依據。
猜你喜歡
保健醫苑(2022年5期)2022-06-10 07:46:18
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
第一財經(2021年6期)2021-06-10 13:19:08
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
Coco薇(2017年9期)2017-09-07 21:23:49
紡織服裝流行趨勢展望(2016年2期)2016-05-04 03:47:15
中國當代醫藥(2015年7期)2015-03-01 02:01:09
汽車科技(2015年1期)2015-02-28 12:14:44
