有機反應中的親核性和堿性
2014-01-26 10:32:48呂萍王彥廣
大學化學 2014年1期
呂萍 王彥廣
(浙江大學化學系 浙江杭州 310027)
有機反應中的親核性和堿性
呂萍 王彥廣
(浙江大學化學系 浙江杭州 310027)
本文系統討論了有機化學教學中經常遇到的親核性與堿性關系的問題,并介紹了親核性和堿性在預測有機反應區域選擇性方面的應用。
親核性 堿性 軟硬酸堿理論 前線分子軌道理論 有機化學教學
親核取代反應、親核加成反應和堿催化的消除反應是基礎有機化學教學的重要內容,因此,掌握試劑的親核性和堿性規律對于學好有機化學至關重要。親核試劑的親核性在許多情況下與堿性的大小順序一致,但在一些情況下卻相反,這往往會給初學者帶來很大困惑。為此,本文試圖從堿和親核試劑的定義出發,通過熱力學和動力學概念闡述堿性和親核性的本質區別,并通過實例介紹親核性和堿性在預測有機反應選擇性方面的應用。
1 基本概念
1.1 酸和堿
有關酸和堿的理論主要有兩種,即Br?nsted酸堿理論和Lewis酸堿理論[1]。按照Br?nsted酸堿理論,酸是質子的給體,堿是質子的接受體;而按照Lewis酸堿理論,酸是電子對的接受體,堿是電子對的給體。與Br?nsted酸堿相比,Lewis酸堿的范圍更廣,幾乎所有離子型的有機反應均可以認為是Lewis酸堿的反應。例如,在烯烴的親電加成中,烯烴在成鍵時給出電子,所以是Lewis堿,親電試劑在成鍵時接受電子,因而屬于Lewis酸;在羰基的親核加成中,親核試劑在成鍵時給出電子,屬于Lewis堿,而羰基化合物在成鍵時接受電子,則是Lewis酸。
1.2 親核試劑和親電試劑
親核試劑(nucleophile)指的是一個原子、離子或分子擁有電子對,具有富電子性,在與缺電性物種反應時,能提供電子對而成鍵;物種所擁有的這種性質稱為親核性(nucleophilicity)。親電試劑(electrophile)指的是一個原子、離子或分子缺少電子對,具有缺電子性,在與富電性物種反應時,能夠接受電子對而成鍵;物種所擁有的這種性質稱為親電性(electrophilicity)。由此看來,親核試劑是Lewis堿,親電試劑是Lewis酸。據此,人們就容易得出“堿性越強親核性就越強”的結論。
1.3 親核性和堿性的相對強弱
當一個富電性物種與質子反應時,會表現出堿性(basicity);而當這個富電性物種親核進攻缺電性碳時,則會表現出親核性。顯然,試劑的親核性和堿性是不同的,而且在反應中有競爭,競爭的結果直接導致了有機反應的化學選擇性。例如,富電性物種試劑在與鹵代烷發生反應時,如果它奪取碳正離子中間體β-碳上的質子,表現出堿性,生成消除產物(圖1,途徑a);如果它與碳正離子結合,則表現出親核性,生成取代產物(圖1,途徑b)。我們可以用堿性較強但親核性較弱的試劑來選擇性地得到消除產物,亦可用親核性較強但堿性較弱的試劑來選擇性地得到取代產物。
如何評價堿性和親核性的強弱?對堿(B)的堿性,常用其共軛酸(HB)的pKa值來評價其強弱。同一分子既可以是酸,也可以是堿。如水分子,它不僅能提供質子,還能接受質子。如果我們用水做標準來評價酸堿性,堿(B-)、水(H2O)、共軛酸(HB)和共軛堿(OH-)之間存在一定的平衡,由此平衡我們可
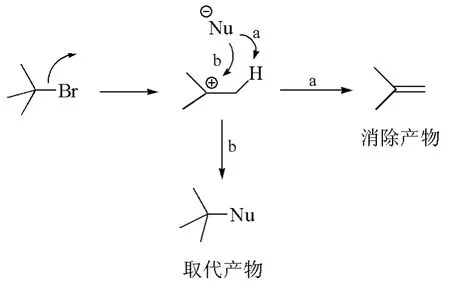
圖1 消除反應與取代反應的競爭途徑
以得出平衡常數Ka和pKa。Ka越大,pKa就越小,表明共軛酸(HB)的酸性越強,堿(B)的堿性越弱,如下所示:
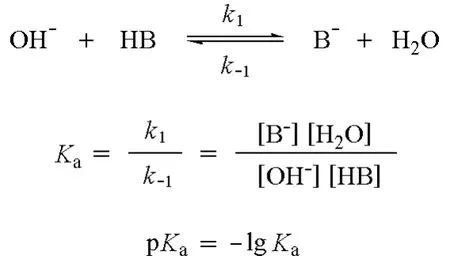
常見堿的pKa數據如表1所示。
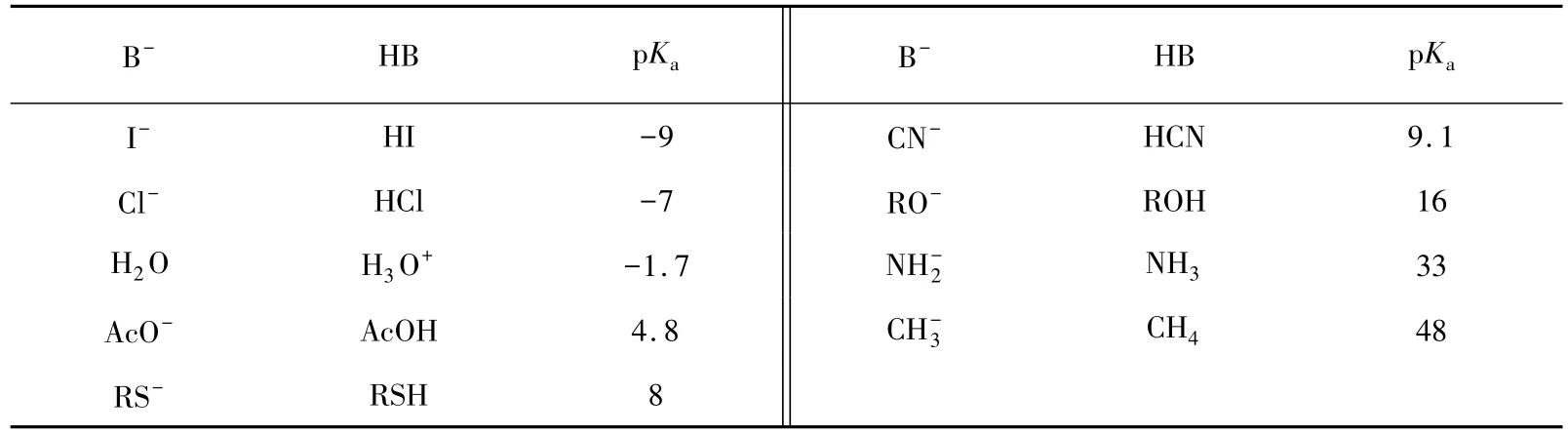
表1 一些堿的共軛酸的pKa數據
最早提出評價親核性強弱是Swain和Scott[2],他們的理論基于溴甲烷在水中的親核取代反應:

將溴甲烷在25℃條件下的水解看成一個標準反應,速度常數為k0,用s代表親電物質對親核試劑的敏感性(標準反應的s=1)。加入一定濃度不同的親核試劑,鹵代烴被親核取代的速度常數為k,用n代表親核性的強弱,也稱親核常數(標準反應的n=0)。由此,鹵代烴與不同親核試劑反應的線性自由能關系可以用Swain-Scott方程來表示:
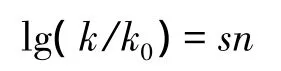
通過動力學跟蹤和計算,固定底物,得出不同親核試劑的親核常數n,數值越大,親核性越強,親核取代反應的速度也就越快。固定親核試劑,則可得出不同的鹵代烴對親核試劑的敏感性s,數值越大,對親核試劑越敏感,親核取代反應的速度也就越快(表2)。
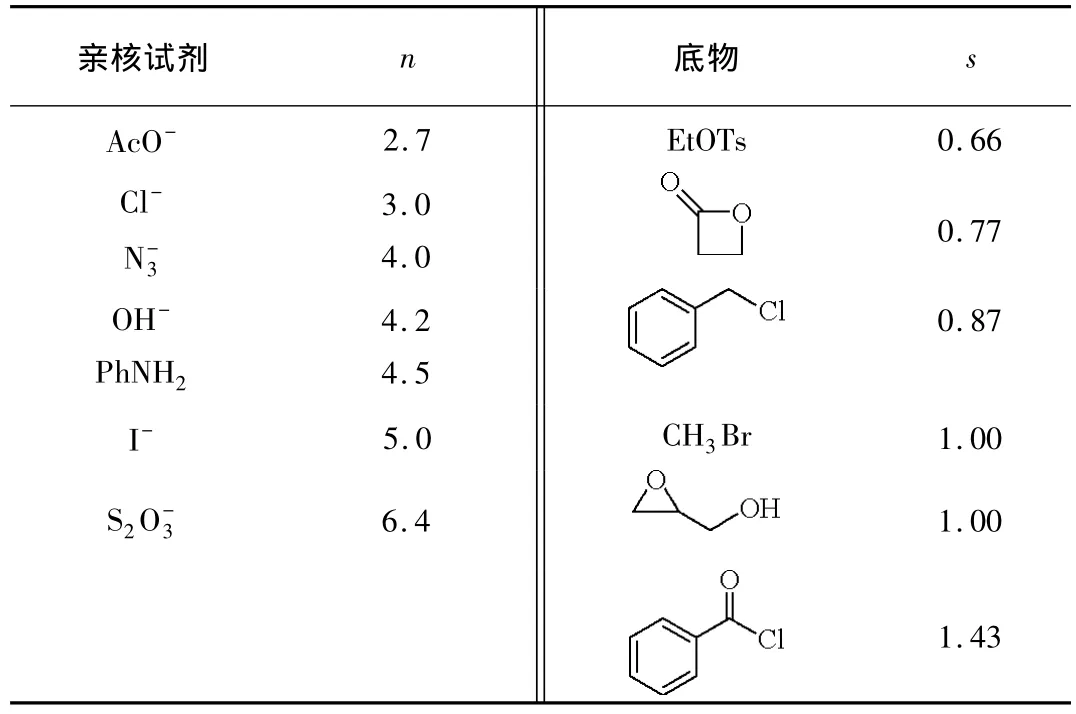
表2 Swain-Scott方程中的參數
評價親核性強弱的方法還有很多,如Ritchie方程[3-4]、Edwards方程[5-6]、Mayr方程等[7-10]。總之,一個富電性物種的親核性可以相對量化,選定一個親核加成或親核取代的模型,就可以對親核性的強弱進行評價。一些常用親核試劑的親核性強弱順序如下:
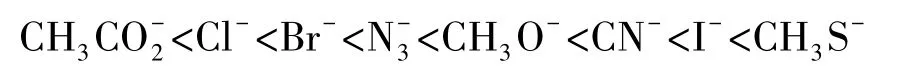
2 親核性和堿性的關系
雖然堿和親核試劑都是給出電子對成鍵,但二者有本質的區別。堿性是基于酸堿平衡的評價,與平衡常數有關;而親核性是基于親核試劑和親電試劑反應性的評價,與反應速度有關。前者是熱力學概念,后者則是動力學概念。從反應進程圖中可以看出這一本質的不同(圖2)。堿性描述的是堿奪得質子前后的相對穩定性(反應放出的熱);親核性描述的是親核試劑和底物的作用能力(反應所需要克服的活化能)。
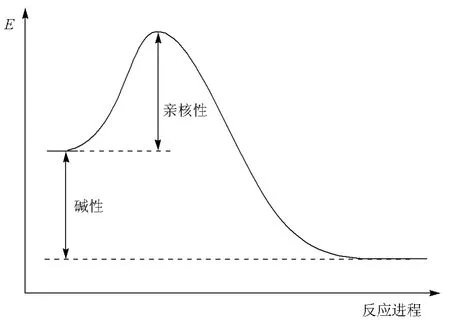
圖2 反應進程中親核性與堿性的關系
由此,我們不難理解為什么表1和表2中親核性與堿性的次序不完全一致,如碘負離子的堿性比氯負離子弱,但它的親核性比氯負離子強。大量研究表明,親核性和堿性的強弱主要有以下規律[11]:
(1)對于親核進攻元素是同一個元素來講,負電性物種的親核性大于中性物種的親核性,如:OH->H2O,NH>NH3。
(2)對于親核進攻元素是同一周期元素來講,親核性和堿性基本一致,如:CH3O->OH->PhO->CH3COO->TsO-;CH>NH>OH->F-。
(3)對于親核進攻元素是同一族元素來講,堿性由上到下依次減弱(即F->Cl->Br->I-),親核性由上到下依次增強(即F-<Cl-<Br-<I-)。
(4)α-效應:當親核原子的鄰位有雜原子時,親核性增強。例如,雖然OH-(共軛酸的pKa=15.7)的堿性比OOH-(共軛酸的pKa=11.6)大,但OH-的溶劑化作用比OOH-強,降低了其親核能力,相應地,OOH-的親核性強。同樣,氨(共軛酸的pKa=9.3)的堿性比肼(共軛酸的pKa=8.0)大,但肼的親核性比胺強。
(5)立體效應:如EtOH、i-PrOH和t-BuOH的pKa分別為16.0、17.1和19.2,它們共軛堿的堿性強弱為t-BuO->i-PrO->EtO-;但它們的親核性受到空間位阻的影響,親核性強弱為EtO->i-PrO->t-BuO-。有趣的是,1,8-雙(二乙基氨基)-2,7-二甲氧基萘是一個很強的堿(其共軛酸的pKa為16.3),被稱為“質子海綿”,但由于高位阻原因,其親核性很弱。
在有機合成中,為了避免親核取代或親核加成負反應發生,常使用一些高位阻的堿來奪取質子,如2,6-二甲基吡啶、2,6-二叔丁基吡啶、二異丙基胺基鋰(LDA)等。
(6)溶劑效應:當親核試劑與溶劑之間發生作用,如圖3所示,質子性溶劑甲醇將陰離子包絡在中心,穩定陰離子,使陰離子失去親核能力;極性非質子性溶劑二甲基亞砜(DMSO)將陽離子包絡在中心,穩定陽離子,陰離子成裸露離子,從而提高了陰離子的親核能力。
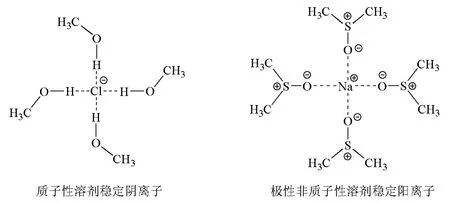
圖3 氯離子和鈉離子的溶劑化作用
在DMSO溶劑中,一些常見親核試劑的親核性強弱順序如下:
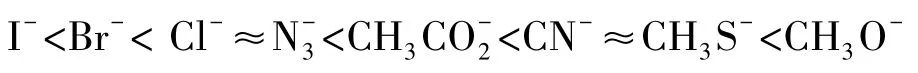
溶劑效應還能改變兩可親核試劑(ambident nucleophiles)反應的區域選擇性。如圖4所示,2-萘酚的鈉鹽與芐基溴反應時,在非質子極性溶劑DMF和DMSO中,鈉離子的強溶劑化作用導致了氧原子的親核性增強,從而取代反應主要生成氧烷基化產物(A)。在水或三氟乙醇等極性質子性溶劑中,由于酚氧負離子的溶劑化作用大大增強,其親核性減弱,結果鄰位碳原子親核取代的產物(B)顯著增加[12]。由此可見,可以通過選擇合適的反應溶劑來調控兩可親核試劑反應的區域選擇性。
必須指出的是,盡管親核性是動力學概念,但一些兩可親核試劑反應的最終結果有時取決于產物的熱力學穩定性。如圖5所示,在1-巰基-8-辛胺與碘甲烷的反應中,巰基的親核性比氨基強,易于進攻碘甲烷;但氨基的堿性比巰基強,而且C—N鍵比C—S鍵穩定,從而有利于生成熱力學穩定的銨鹽。在這個過程中,氨基最終被甲基化,而巰基相當于一個催化劑。
有些兩可親電試劑(ambident electrophiles)的反應性同樣取決于產物的熱力學穩定性。如圖6所示,α,β-不飽和酮與CN-發生親核加成時,在低溫時主要生成動力學控制的1,2-加成產物,升高溫度則有利于生成熱力學控制的1,4-加成產物[13]。
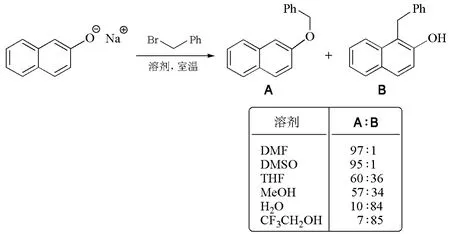
圖4 溶劑效應對反應區域選擇性的影響
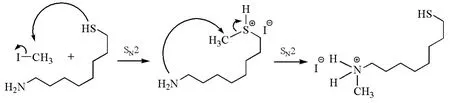
圖5 熱力學控制的硫醇胺的區域選擇性親核取代
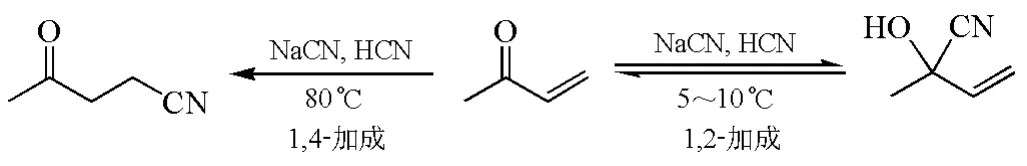
圖6 熱力學控制的α,β-不飽和酮的區域選擇性親核加成
3 親核性的理論解釋
不論是親核取代還是親核加成,對親核性相對強弱的解釋可以歸納為兩種,一種是Klopman-Salem理論[14-16],另一種是軟硬酸堿理論[17-21]。
3.1 Klopman-Salem理論
Klopman-Salem提出有機反應受3個因素制約[22]:閉殼層排斥力(the closed-shell repulsion)、庫侖或靜電相互作用力和電荷轉移能力。閉殼層排斥力指的是分子或離子有很多填充在滿軌道中的電子,這些電子之間的排斥導致分子或離子間的排斥,最終導致分子或離子的碰撞并不都是有效碰撞;只有當分子或離子有足夠的能量克服反應的活化能,才是真正的有效碰撞,才能發生反應。庫侖或靜電相互作用力指的是極性分子或離子有一定的偶極矩,它們靠近的時候是有方向性的,帶正電的和帶負電的相互吸引而發生反應。電荷轉移能力指的是富電性物種的HOMO和缺電性物種的LUMO之間的相互作用能。
當親核進攻的原子相同時,可以用靜電作用解釋親核性的相對強弱。如上述有關氧作為反應中心的親核性強弱:CH3O->OH->PhO->CH3COO->TsO-。在這個序列中,甲氧基中的氧原子擁有的電荷密度最大,而對甲苯磺酸根陰離子中的氧原子擁有的電荷密度最小,由此從前往后親核能力降低。這與堿性的次序是一致的。
當親核進攻原子不同時,可以用親核試劑LUMO和底物HOMO之間的相互作用來解釋。如圖7所示,硫代乙酸根陰離子有兩個共振結構(Ⅰ和Ⅱ),進攻伯鹵代烴可分別得到硫取代產物和氧取代產物。事實證明,硫的親核性強,反應能選擇性地得到硫取代產物。用電荷轉移能力來解釋,就是硫孤對電子所占據的非鍵軌道n和鹵代烴中反鍵軌道σ*能量間隙小,容易給出電子而成鍵(圖8)[23]。
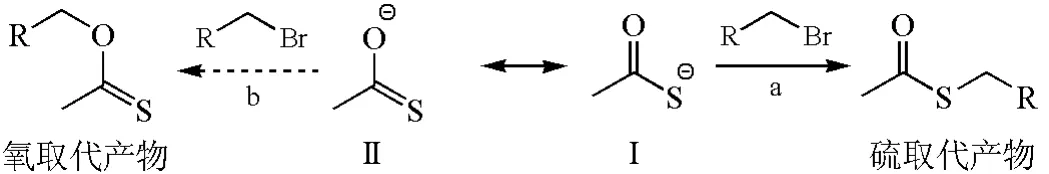
圖7 硫代乙酸根陰離子與溴代烷的選擇性親核取代
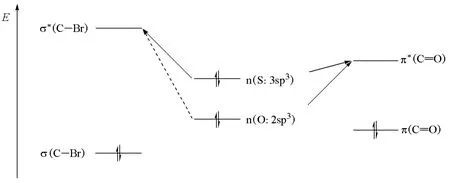
圖8 硫代乙酸根陰離子親核取代反應的電荷轉移能力示意圖
當缺電性物種是羰基時,由于羰基的π*軌道比鹵代烴的σ*軌道能量低(圖8),此時由于氧的n軌道與羰基的π*軌道能量間隙減小,硫和氧都能作為親核試劑,故羰基對硫和氧的選擇性要低得多。
3.2 軟硬酸堿理論
Pearson提出了軟硬酸堿(HSAB)理論,根據IUPAC定義,“軟酸”指的是一個缺電子的、可以接受電子的Lewis酸,它具有易極化的性質;相應地,“軟堿”指的是一個富電子的、可以給出電子的Lewis堿,它具有易極化的性質。軟硬酸堿理論認為:軟酸易與軟堿作用,即“軟親軟”;而硬酸易與硬堿作用,即“硬親硬”。雖然文獻中也有關于軟硬度的評價,但HSAB理論僅限于定性判斷化合物的穩定性或反應的可行性。
具體來講,硬酸和硬堿具有小尺寸、高氧化態、低極化、大的電負性等特性,作為硬堿具有低的HOMO軌道能級,作為硬酸具有高的LUMO軌道能級;軟酸和軟堿具有大尺寸、低氧化態、高極化、小的電負性等特性,作為軟堿比硬堿有更高的HOMO軌道能級,作為軟酸比硬酸有更低的LUMO軌道能級。一些常見離子或分子的軟硬酸堿分類列于表3。
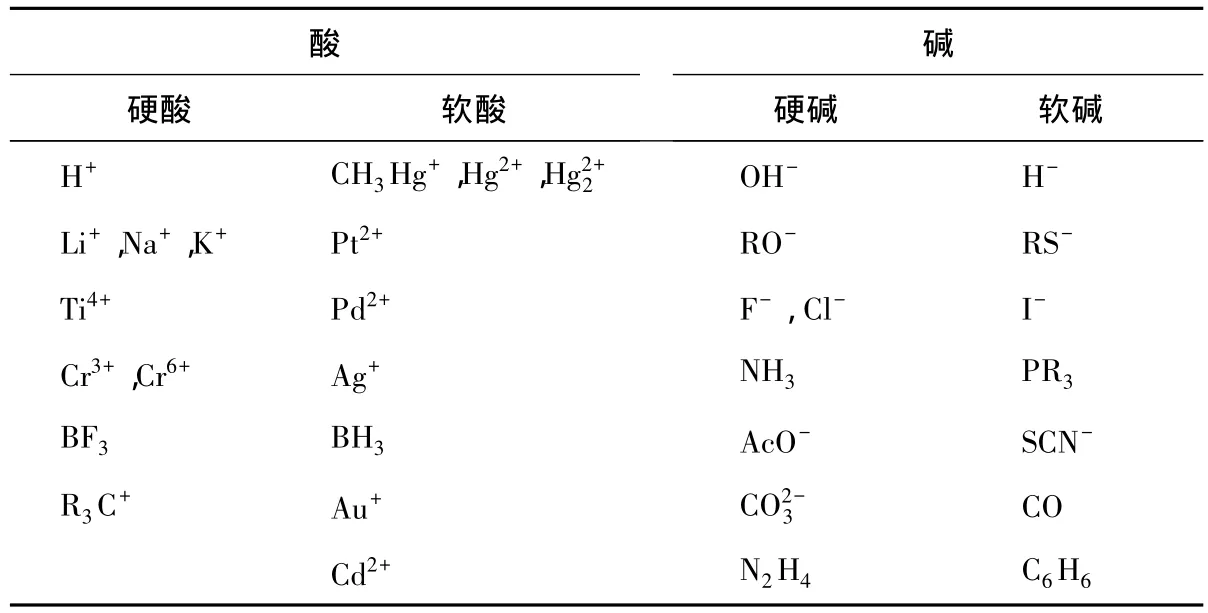
表3 一些常見的軟硬酸堿
與軟硬酸堿相對應,親核試劑也分為硬親核試劑和軟親核試劑,處于兩者之間的稱“邊界”親核試劑,列于表4。反應時,硬親核試劑和硬親電試劑受靜電作用控制而成鍵,軟親核試劑和軟親電試劑受軌道作用控制而成鍵。
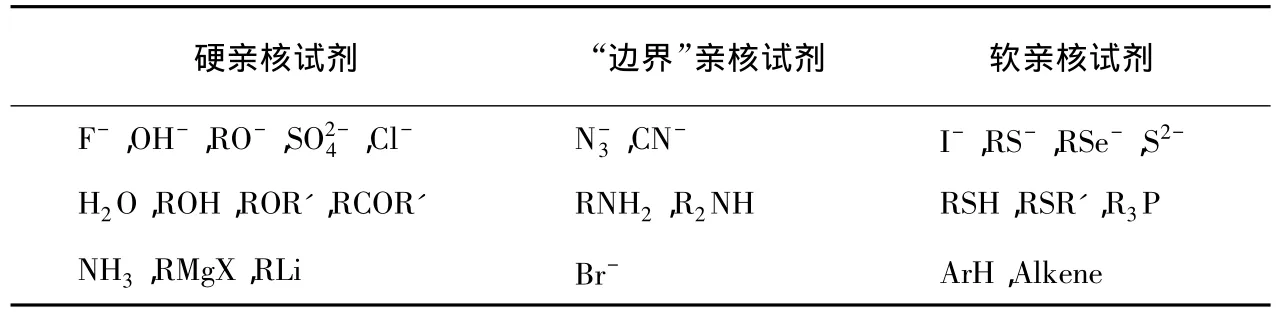
表4 一些常見的軟硬親核試劑
很多兩可親核試劑在進行親核反應時具有一定的區域選擇性,這種選擇性可用HSAB理論來解釋。如圖9所示,在SN1反應中,較硬的親核試劑有利于進攻較硬的親電試劑,即碳正離子;而在SN2反應中,較軟的親核試劑有利于進攻較軟的帶部分正電荷的碳原子[11]。
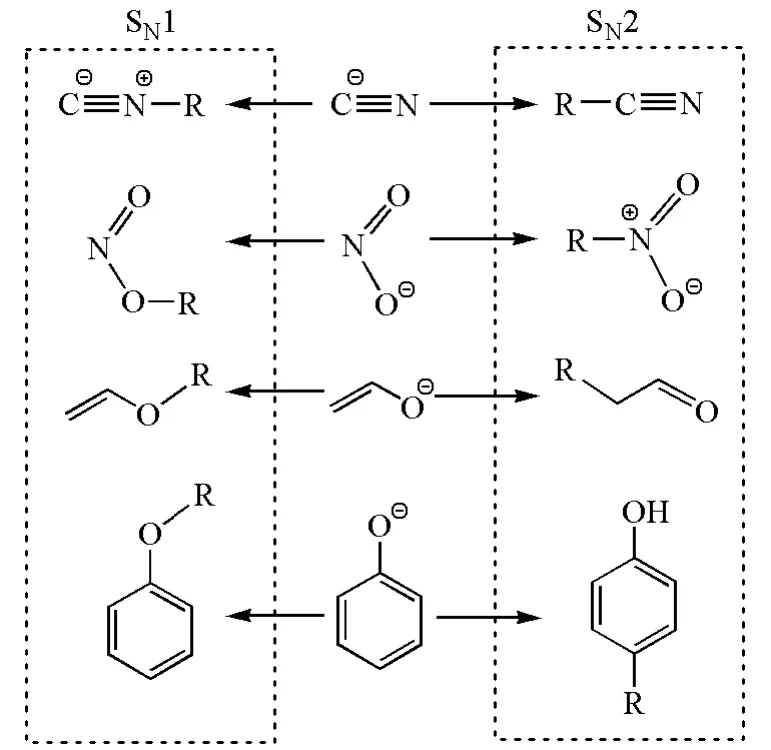
圖9 一些兩可親核試劑的區域選擇性親核取代反應
對于典型的兩可離子CN-來說,當NaCN與碘代烷反應時,CN-的碳原子(軟親核試劑)進攻軟的親電試劑碘代烷,得到腈(圖10)。然而,當使用AgCN時,CN-的氮原子(硬親核試劑)傾向于與碘代烷產生的碳正離子“硬親硬”,生成異腈[24]。
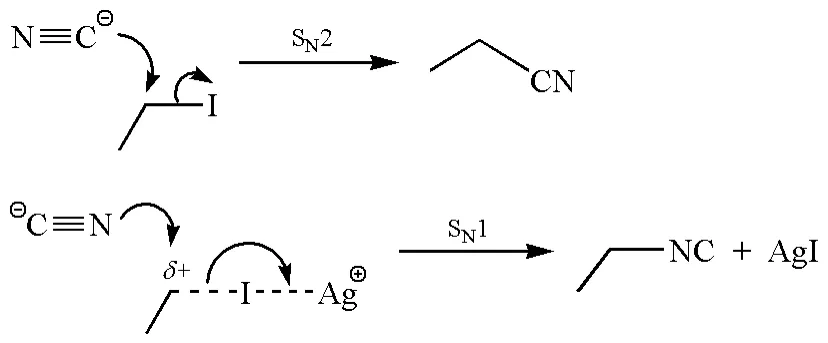
圖10 碘代烷與NaCN或AgCN反應的區域選擇性
HSAB理論還可解釋一些兩可親電試劑的反應性。α,β-不飽和羰基化合物是典型的兩可親電試劑,其α-C和β-C相比較,α-C比較“硬”,是硬親電試劑,與有機鋰、Grignard試劑等硬親核試劑作用時,受靜電作用的控制,容易發生1,2-加成;β-C比較“軟”,與軟親核試劑作用時,受軌道的控制,容易發生共軛加成。例如,α,β-不飽和醛與烷基鋰試劑反應主要發生1,2-加成,而與硫醇反應則主要發生1,4-共軛加成(圖11)。
最近的一個例子是α-二鹵代砜(PhSO2CX2H)在強堿LiN(SiMe3)2(LHMDS)存在下與α,β-不飽和酮的親核加成反應[25]。反應中所形成的碳負離子PhSO2CF是較“硬”的堿,而PhSO2CCl是較“軟”的堿,因此PhSO2CF與α,β-不飽和酮反應生成1,2-加成產物,PhSO2CCl則生成1,4-加成產物(圖12),體現了“軟親軟”、“硬親硬”規律。
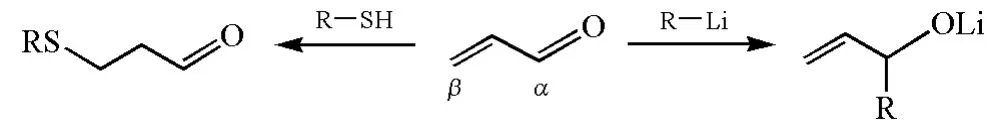
圖11 α,β不飽和醛與烷基鋰或硫醇反應的區域選擇性
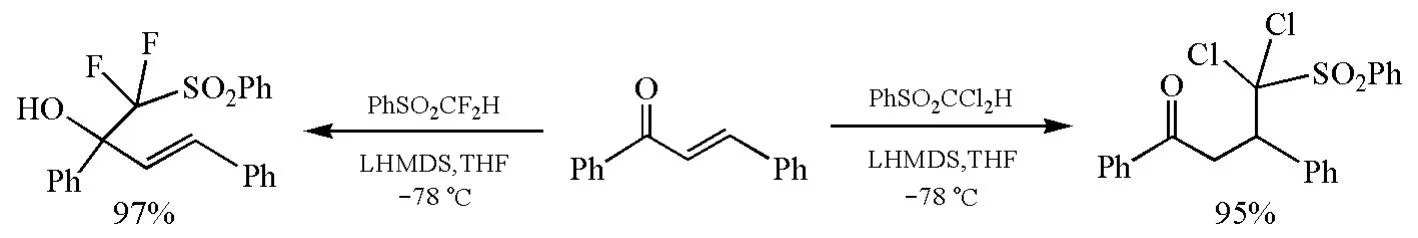
圖12 α,β-不飽和酮與PhSO2CF或PhSO2CC反應的區域選擇性
4 結束語
親核性和堿性是富電性分子或離子所具有的特性,二者既有共性,又有本質區別。盡管人們對堿性的相對強弱已能夠用其共軛酸的pKa進行定量描述,但迄今尚未找到可被普遍應用的定量描述親核性相對強弱的參數。在有機反應過程中,試劑最終體現親核性還是堿性,一般取決于其電子效應、立體效應、溶劑化作用等多種因素。HSAB理論在解釋一些兩可親核試劑反應的區域選擇性方面比較成功,但仍有很多現象難以用該理論進行解釋[24]。
[1]Muller P.Pure Appl Chem,1994,66:1077
[2]Swain C G,Scott C B.J Am Chem Soc,1953,75:141
[3]Ritchie C D.Acc Chem Res,1972,348
[4]Ritchie C D.J Am Chem Soc,1975,1170
[5]Edwards J O.J Am Chm Soc,1954,76:1540
[6]Edwards J O.J Am Chem Soc,1956,78:1819
[7]Mayr H,Bug T,Gotta M F,et al.J Am Chem Soc,2001,123:9500
[8]Mayr H,Ofial A R.Pure Appl Chem,2005,77:1807
[9]Phan T B,Breugst M,Mayr H.Angew Chem Int Ed,2006,45:3869
[10]Mayr H,Ofial A R.J Phys Org Chem,2008,21:584
[11]Smith M B,March J.March's Advanced Organic Chemistry,Reactions,Mechanisms,and Structure.6th ed.New York:John Wiley&Sons Inc,2007
[12]Kornblum N,Seltzer R,Haberfield P.J Am Chem Soc,1963,85:1148
[13]Clayden J,Greeves N,Warren S,et al.Organic Chemistry.Oxford:Oxford University Press,2001
[14]Klopman G.J Am Chem Soc,1968,90:223
[15]Salem L.J Am Chem Soc,1968,90:543
[16]Fleming I.Frontier Orbitals and Organic Chemical Reactions.Chichester:Wiley,1976
[17]Pearson R G.J Am Chem Soc,1963,85:3533
[18]Pearson R G.Science,1966,151:172
[19]Pearson R G,Songstad J.J Am Chem Soc,1967,89:1827
[20]Pearson R G.J Chem Edu,1968,45:581
[21]Pearson R G.J Chem Edu,1968,45:643
[22]Stone A J,Erskine R W.J Am Chem Soc,1980,102:7185
[23]Clayden J,Greeves N,Warren S,et al.Organic Chemistry.Oxford:Oxford University Press,2009
[24]Mayr H,Breugst M,Ofial A R.Angew Chem Int Ed,2011,50:6470
[25]Ni C F,Zhang L J,Hu J B.J Org Chem,2008,73:5699
