靶向抑制DNA拓撲異構酶的抗腫瘤藥物研究
2014-09-18 08:43:30錢晨彭再利巢暉
大學化學 2014年1期
關鍵詞:研究
錢晨 彭再利 巢暉
(中山大學化學與化學工程學院 廣東廣州510275)
無論在原核或真核細胞中,天然狀態的DNA都主要以超螺旋的形式存在。在DNA的轉錄、復制以及基因表達過程中,DNA雙螺旋鏈需要在超螺旋狀態與解旋狀態之間不斷進行轉換。為了解決DNA在代謝過程中出現的構象問題,生物在長期進化的過程中演化出了DNA拓撲異構酶(topoisomerase,Topo),可通過催化作用改變DNA的拓撲結構[1]。研究發現,與正常細胞不同,拓撲異構酶在腫瘤細胞中表現出不受其他因素影響的高水平表達。因此抑制DNA拓撲異構酶的活性就能阻止腫瘤細胞快速增殖,進而誘導腫瘤細胞的凋亡及壞死。近年來,以DNA拓撲異構酶為靶點設計各類抑制劑,用作抗癌藥物的研究已成為腫瘤化療的重要途徑。本文將對拓撲異構酶的結構、功能及以其為靶點的抗腫瘤藥物研究進展進行介紹。
1 DNA拓撲異構酶的結構與功能
拓撲異構酶是通過兩個連續的轉酯化反應重復斷開和連接DNA主鏈的磷酸二酯鍵來催化DNA拓撲結構轉變,進而直接參與或影響細胞DNA的復制、翻譯、轉錄、重組以及有絲分裂等生命過程[2]。根據拓撲異構酶催化機制的不同,主要將其分為兩類:TypeⅠ拓撲結構酶與 TypeⅡ拓撲結構酶[3]。TypeⅠ拓撲結構酶屬于單體酶,包括TopoⅠA、TopoⅠB和TopoⅠC 3種亞型,它在作用過程中不需要額外能量因子。TypeⅡ拓撲結構酶是同源二聚體酶,包括TopoⅡα、TopoⅡβ兩種亞型[4]。在人細胞中,主要包含DNA拓撲異構酶Ⅰ(TopoⅠ)和DNA拓撲異構酶Ⅱ(TopoⅡ)兩種,分別歸類于TopoⅠB和 TopoⅡα。
1.1 TopoⅠ的結構與功能
TopoⅠ最早是1971年在細菌中發現的,人類TopoⅠ是哺乳動物TopoⅠ的原始型,相對分子質量為100 000,位于染色體20q12~13.2單拷貝基因編碼上。TopoⅠ是一種單體酶(圖1a),由756個氨基酸殘基組成,其中起催化作用的酪氨酸(Tyr)位于羧基端。TopoⅠ在DNA雙鏈上產生單鏈斷裂,使另一單鏈從缺口處穿過,改變DNA超螺旋或螺旋化不足的情況。TopoⅠ中的Tyr與DNA 3'斷端的磷酸鹽以共價鍵結合,同時在斷裂缺口的5'位形成羥基末端(圖1b),5'羥基末端繞另一完整的DNA鏈旋轉,此過程具有可逆性,不需要ATP和二價金屬陽離子的參與。這種結構上的特性使TopoⅠ對正超螺旋和負超螺旋DNA具有幾乎相同的松弛能力[5]。
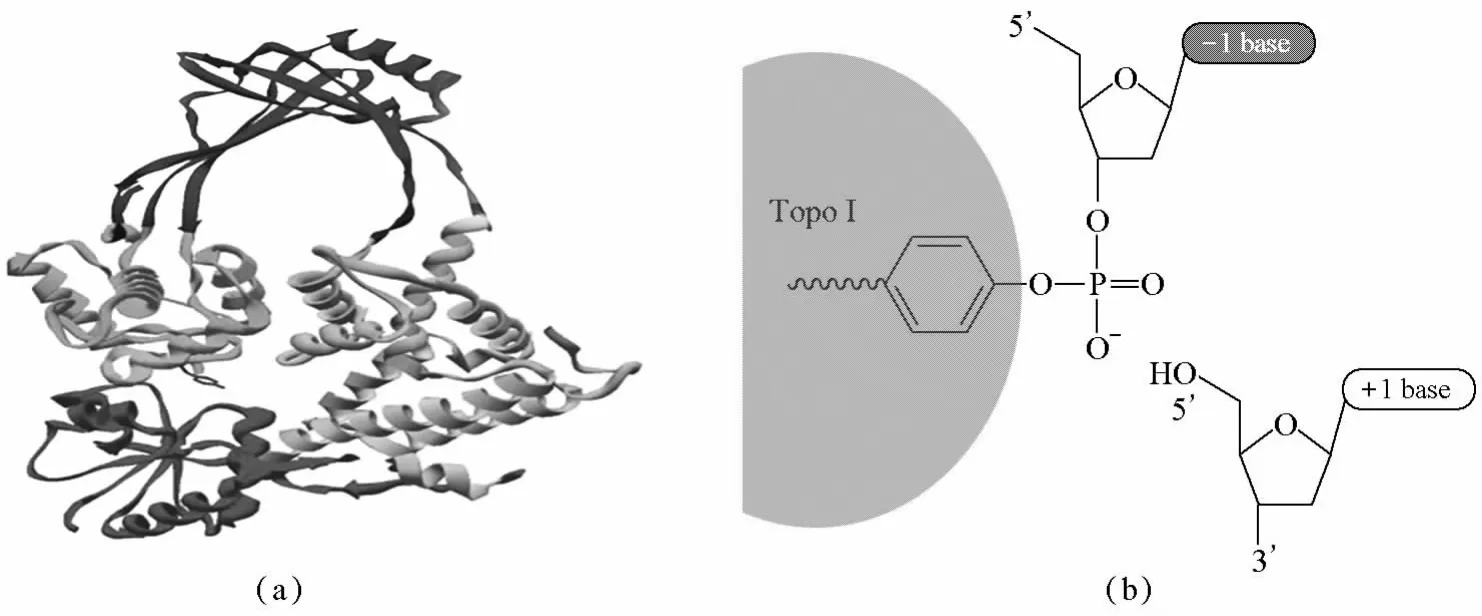
圖1 TopoⅠ酶的結構(a)及TopoⅠ酶與DNA的結合位點示意圖(b)
TopoⅠ催化單鏈斷裂和連接的過程如圖2[2]所示:第一步,酶識別結合到特異DNA序列上;第二步,切開單鏈,酶通過磷酸二酯鍵與DNA共價結合形成可斷裂復合物;第三步,酶在斷裂單鏈上的過渡;第四步,在切開位點上對斷裂單鏈進行重連;第五步,繼續解旋,重復應用活化后的可斷裂復合物;第六步,解旋之后,酶從DNA上釋放出來[6]。

圖2 TopoⅠ作用機理圖
1.2 TopoⅡ的結構與功能
TopoⅡ最早發現于1980年,人類TopoⅡ也是哺乳動物TopoⅡ的原始型,位于人染色體17q21~22單拷貝基因編碼的二聚體蛋白上,表達依賴于細胞周期。TopoⅡ是由兩個相同亞基組成的二聚體(圖3a)。TopoⅡ在DNA主鏈上產生雙鏈斷裂,使另一條雙鏈DNA從缺口處穿過,在整個過程中需要ATP提供能量。除了能完成所有TopoⅠ的功能以外,TopoⅡ還能在DNA復制完成后分開相互交聯的姐妹染色單體。TopoⅡ能對特殊的DNA序列進行識別,并通過Tyr與DNA 5'斷端的磷酸基以共價鍵結合(圖3b)[5],形成易解離復合物,改變DNA的拓撲結構,然后再催化斷裂鏈的重新連接[7]。
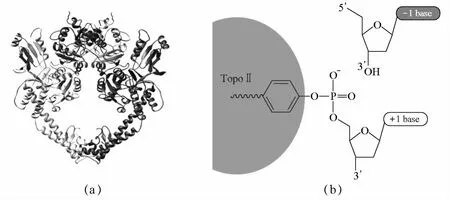
圖3 TopoⅡ酶的結構(a)及TopoⅡ酶與DNA的結合位點示意圖(b)
TopoⅡ可以剪切和打開一股雙螺旋鏈,其催化過程如圖4[2]所示:第一步,酶識別結合到特異DNA序列上;第二步,切開雙鏈,酶與DNA斷裂后產生的5'端結合,形成酶-DNA共價復合物;第三步,另一完整的雙鏈DNA穿過主鏈切開的位點,此過程需結合ATP;第四步,在切開位點上對雙鏈進行重連;第五步,與酶結合的ATP酶水解;第六步,酶周轉,回到能重新啟動催化反應的狀態[8]。

圖4 TopoⅡ作用機理圖
2 DNA拓撲異構酶抑制劑作為抗腫瘤藥物的研究
從細菌到人類,拓撲異構酶廣泛存在于各種生物體的細胞中。與正常細胞不同的是,拓撲異構酶在腫瘤細胞中均表現出不受其他因素影響的高水平表達,盡管原因尚不清楚,但這卻是拓撲異構酶抑制劑對腫瘤細胞具有特殊療效的細胞學基礎。近年來,人們發現許多抗腫瘤藥都是通過拓撲異構酶發揮療效的。因此,以DNA拓撲異構酶為靶分子設計各種抑制劑,并使其成為抗腫瘤藥物,已成為腫瘤化療研究新熱點。
2.1 拓撲異構酶抑制劑的作用機理
現今研究業已證實,可通過影響Topo酶作用過程的各個階段來破壞酶的活性。既可以直接作用于DNA,也可以作用于拓撲異構酶,還可以作用于DNA拓撲異構酶-DNA斷裂復合物,來完成對拓撲異構酶活性的抑制,并最終導致細胞凋亡[9]。抑制劑的作用實際上是使細胞內功能正常的拓撲異構酶轉變為導致DNA鏈斷裂的致傷物,而細胞死亡的最終原因可能是由于DNA鏈斷裂的錯誤修復或是由于可斷裂復合物的形成及穩定存在,激活了細胞內一系列導致細胞程序性死亡的過程。
大體上來說,DNA拓撲異構酶抑制劑的抑制機理可以分為兩種,一種是毒性機理,一種是催化抑制機理。
毒性機理是指抑制劑與Topo-DNA共價復合物形成三元復合物。通過提高Topo-DNA共價復合物的穩態濃度使Topo酶“中毒”。具體地說,在正常情況下,拓撲異構酶能使DNA發生瞬間斷裂和重接,但是在藥物作用下,TopoⅠ或TopoⅡ在切開單鏈或雙鏈后,拓撲異構酶與末端DNA以共價鍵形成一種易解離的復合物(酶-DNA可斷裂復合物)(圖5)[3],并使這一可逆的平衡反應趨向于酶-DNA可斷裂復合物一側,從而導致DNA鏈的斷裂,并進一步啟動一系列導致細胞死亡的因素。
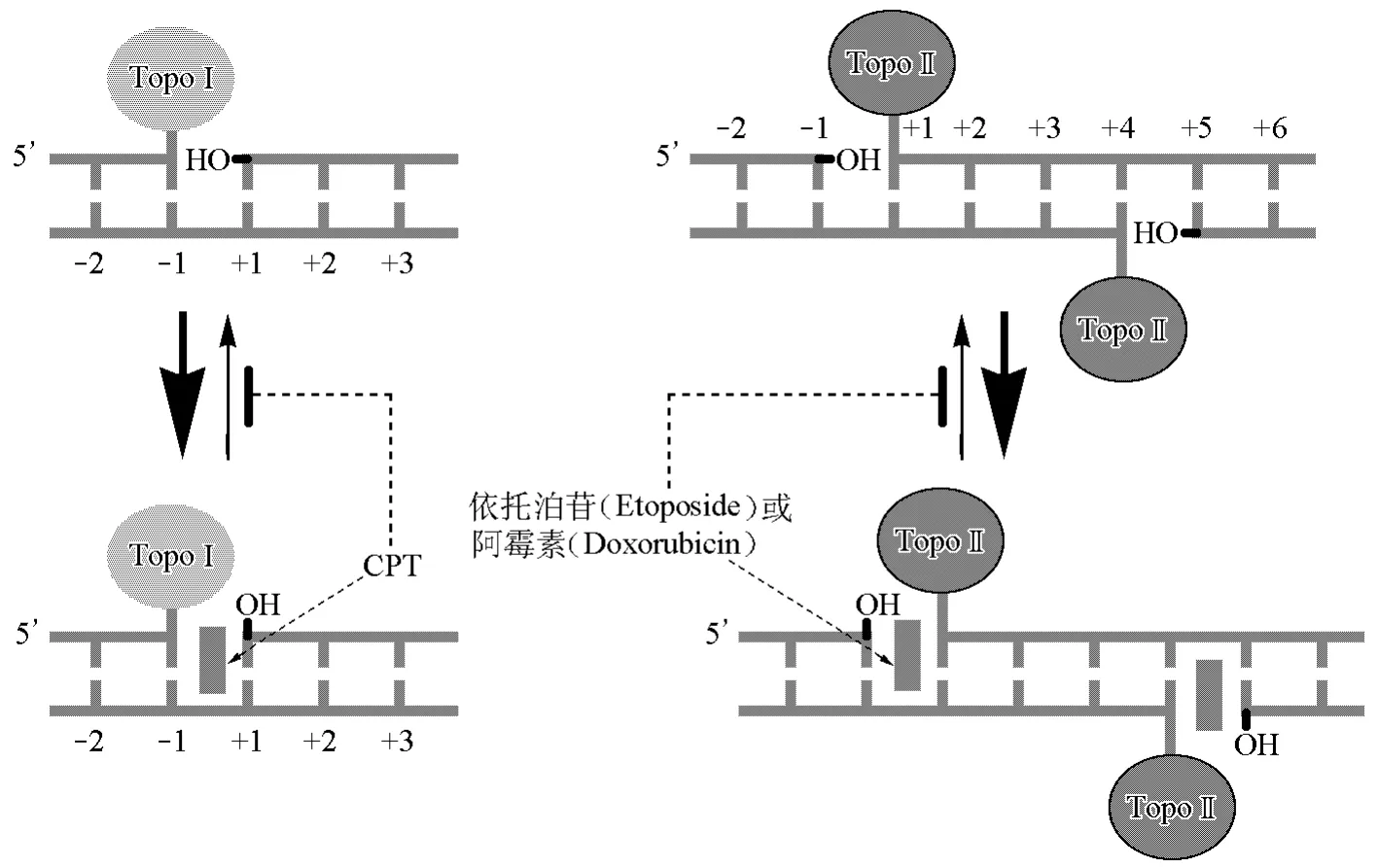
圖5 TopoⅠ/TopoⅡ在拓撲異構酶抑制劑作用下與DNA形成可斷裂復合物
與毒性機理不同,催化機理是指抑制劑通過阻滯Topo的某一特定功能或催化反應中的某一步驟,進而抑制Topo總的催化活性。Topo催化抑制劑主要是一些DNA或蛋白酶的結合劑,包括插入劑如阿克拉霉素A和大小溝結合劑如Hoechst33258等,Topo催化抑制劑可以作用在拓撲異構酶催化反應的各步驟,因此其作用機制也不相同。大致可分為3類:①抑制Topo與DNA的結合,通過阻斷酶與其底物DNA的接觸,化合物可以削弱酶在催化和構象上的功能,如阿柔比星。②穩定Topo-DNA共價復合物,抑制Topo的催化功能,如ICRF-187。③ 阻止ATP鍵合(僅TopoⅡ),如新生霉素[10]。
2.2 有機小分子作為DNA拓撲異構酶抑制劑的研究進展
目前對于DNA拓撲酶抑制劑的研究大部分集中在一些生物堿類有機化合物。根據其作用的底物不同,Topo酶抑制劑可以分為TopoⅠ、TopoⅡ抑制劑和TopoⅠ/Ⅱ雙重抑制劑。
以TopoⅠ為靶點的抗腫瘤藥物主要是喜樹堿及其衍生物[11]。喜樹堿(camptothecin,CPT)類化合物是從琪桐科植物喜樹中分離得到的生物堿,具有較強的細胞毒性,對肺癌、胃癌、腸癌、卵巢癌、膀胱癌及白血病等均有較好療效;但由于CPT內酯環在生理條件下的不穩定性(圖6)及其毒副作用,此類抑制劑的研究重點主要集中在通過對母體CPT的化學結構進行改造,尋求更好的CPT衍生物。目前已有3種CTP類藥物在國內外上市,在臨床上用于結腸癌、卵巢癌和肺癌等的治療;還有30多個CPT衍生物正處于臨床試驗階段。另外,尋找水溶性更好、細胞毒性更強的CPT衍生物的研究也在持續進行[12-13]。CPT類化合物的作用機制是在TopoⅠ作用過程中與DNA形成可斷裂復合物,阻礙DNA鏈的閉合,導致細胞DNA單鏈斷裂的累積。這種單鏈斷裂對細胞來說并不是致死性的,當可斷裂復合物與正在進行復制的DNA復制叉相遇時,會繼發地造成不可逆的DNA雙鏈斷裂,最終引起細胞死亡(圖7)[3]。
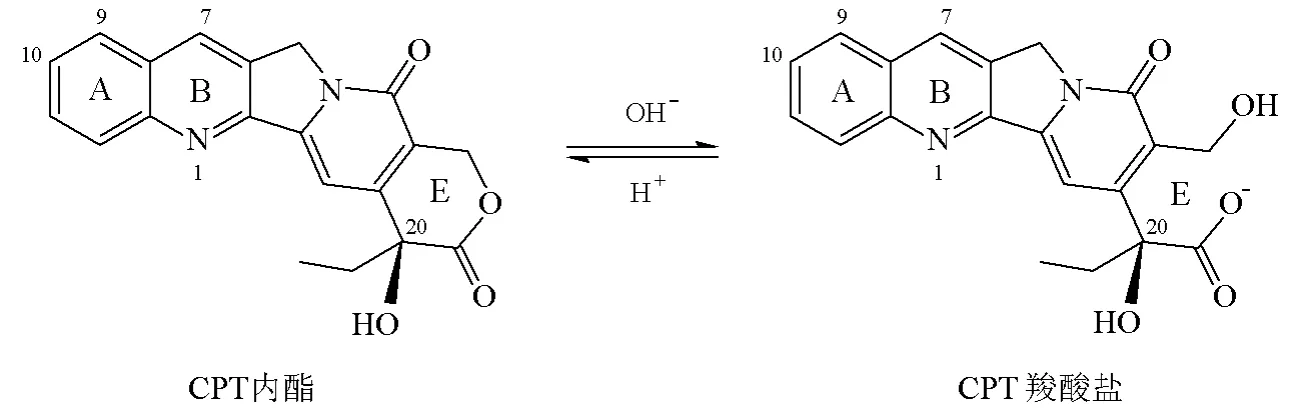
圖6 CPT在生理條件下的不穩定性
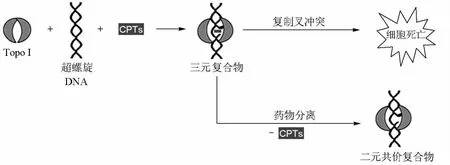
圖7 CPT類TopoⅠ抑制劑的作用機制
CPT類抑制劑和TopoⅠ-DNA形成的三元復合物在藥物濃度降低以后,會很快被逆轉,細胞膜上的轉運蛋白將CPT類化合物轉運出細胞從而產生耐藥性。因此,增強CPT-TopoⅠ-DNA三元復合物的穩定性對此類化合物藥效的發揮有著重要意義。支志明等[14]通過分子動力學研究,提出了預測三元復合物穩定性的方法,并總結了具有穩定作用的CPT衍生物的結構特征。
以TopoⅡ為靶點的抗腫瘤藥物較多,根據與底物的作用方式不同,將TopoⅡ抑制劑分為TopoⅡ毒劑和TopoⅡ催化抑制劑。毒性機理是指抑制劑與TopoⅡ-DNA形成三元復合物,通過提高復合物的穩態濃度使Topo酶“中毒”,受藥細胞基因組DNA產生雙鏈斷裂并累積,從而導致細胞變異或死亡。迄今發現的TopoⅡ抑制劑大部分為TopoⅡ毒劑,常見的有阿霉素 (Doxorubicin),VP-16(Etoposide),沙爾威辛等。與毒性機理不同,催化抑制機理是指抑制劑通過阻滯TopoⅡ的某一功能或催化反應中的某一特定步驟,進而抑制TopoⅡ總的催化活性。此類藥物包括:新生霉素(Novobiocin)、美巴龍(Merbarone)、阿柔比星(Aclarubuxin)、ICRF-193 等[15](圖8)。
研究表明,處于S期的細胞TopoⅡ的含量比較高。TopoⅡ毒劑主要在S期對DNA產生不可逆的雙鏈斷裂,最終導致DNA斷裂復合物在G2期的積累。TopoⅡ催化抑制劑(如阿柔比星)也主要作用于S期,使超螺旋DNA的松弛在DNA復制前減慢,影響G1期到S期的進程,最終導致細胞周期在G2期停滯[16]。吖啶類化合物作為抗菌、抗癌藥物的研究取得了很大進展,而其抗癌作用的原理就在于抑制了TopoⅡ這一關鍵酶。John R.Goodell等[17]通過研究一系列吖啶類化合物對細胞周期的影響,對該系列化合物作為TopoⅡ抑制劑的抑制機理作出了區分(圖9)。

圖8 一些常見的TopoⅡ抑制劑
現今,人們在研究以TopoⅠ或TopoⅡ為靶點的抗癌藥物方面有了長足的進步,然而以單一拓撲酶為靶點的抗癌藥物卻存在著很多缺陷。有研究證明,由TopoⅡ抑制劑造成的DNA雙鏈斷裂會導致二次惡性腫瘤的發生[18];另外,選擇性地抑制TopoⅠ會引起體內TopoⅡ含量上升;并且,TopoⅠ和TopoⅡ抑制劑聯合用藥會引起嚴重的腫瘤細胞多藥耐藥性,甚至會使兩種抑制劑之間發生競爭性抑制,從而降低兩種抑制劑各自的藥效[19]。TopoⅠ和TopoⅡ雙重抑制劑對TopoⅠ和TopoⅡ都有抑制效果,有望解決單一的拓撲酶抑制劑在供藥耐受性上的問題,而且它們可以完全抑制DNA、RNA的合成,進而導致細胞死亡。由于它們的抑制能力一般都比較強,可以減小供藥濃度太大所引起的毒性問題。

圖9 吖啶類TopoⅡ毒劑和TopoⅡ催化抑制劑
大部分TopoⅠ/TopoⅡ雙重抑制劑是TopoⅠ和TopoⅡ的雙重毒劑,能同時穩定兩種拓撲酶與DNA形成的可斷裂復合物,從而抑制拓撲酶的活性。例如近幾年發現的吩嗪類衍生物XR11576,研究證明它能同時介導TopoⅠ/TopoⅡ-DNA可斷裂復合物的形成,是TopoⅠ和TopoⅡ的雙重毒劑,并且這種藥物對過量表達P-糖蛋白的多藥耐藥性腫瘤細胞有很好的殺傷效果,現已進入臨床前試驗階段。另一種研究比較透徹的TopoⅠ/TopoⅡ雙重毒劑茚托利(intoplicine)的作用方式稍有不同,它是通過deep intercalation mode和outside binding mode兩種模式分別穩定了TopoⅠ及TopoⅡ與DNA形成的可斷裂復合物[20]。
近些年來,TopoⅠ/TopoⅡ雙重抑制劑引發了人們日益濃厚的興趣,一系列不同結構、不同作用機制的雙重抑制劑被合成了出來,包括靈菌紅素(Prodigiosin),姜黃素(Curcumin),吲哚基喹啉衍生物TAS-103等(圖10),其中PZA和茚托利辛已進入臨床一期試驗階段[21]。然而,目前關于這些化合物的構效關系以及具體作用機理的研究仍不透徹,這大大制約了這類化合物的應用。至今仍無TopoⅠ/TopoⅡ類雙重抑制劑抗癌藥物進入市場。

圖10 常見的TopoⅠ/TopoⅡ雙重抑制劑
2.3 金屬配合物作為DNA拓撲異構酶抑制劑的研究進展
目前對于DNA拓撲酶抑制劑的研究主要集中在一些生物堿類有機化合物,金屬配合物抑制DNA拓撲異構酶的研究還較少有人涉及。有機類拓撲異構酶抑制劑存在結構復雜、特異性不高、溶解性差、毒性較大等缺點。與有機化合物相比,金屬配合物分子結構具有更好的可塑性,容易在配體上引入其他分子活性基團,可以針對不同的底物結合環境進行相應的結構修飾;而且其豐富的光電磁性質將有助于探索某些復雜的生命過程。
自從20世紀80年代,Barton等在研究金屬配合物與DNA作用時發現Zn2+,Co3+,Ru2+等配位飽和的八面體手性配合物具有識別DNA二級結構的能力,金屬配合物與DNA的識別和結合已成為近年來生物無機化學領域較活躍的研究課題之一。雖然發現不少金屬配合物具有識別和斷裂DNA功能,但真正在DNA拓撲異構酶抑制方面的具體應用還非常少,至今僅有為數不多的關于金屬配合物抑制DNA拓撲異構酶的研究報道,主要集中在鉑類、釕類、金類等金屬配合物方面。
2.3.1 鉑類配合物作為拓撲異構酶抑制劑
鉑類配合物作為研究最早的抗腫瘤藥物,一直備受研究者關注。2004年,J.H.M.Schellens等研究發現由于順鉑與DNA堿基共價結合,當與喜樹堿聯合應用時,喜樹堿的TopoⅠ抑制活性明顯增強[22]。A.H.J.Wang課題組報道了一系列含tpy平面配體的鉑配合物[23],該系列鉑配合物是一類有效的TopoⅠ/Ⅱ 雙重抑制劑,對TopoⅠ/Ⅱ抑制IC50分別達到了10μmol/L與5μmol/L(圖11a)。支志明課題組最近也報道了一系列含修飾tpy配體的鉑配合物(圖11b)[24-25],實驗顯示,該系列鉑配合物是TopoⅡ抑制劑;經過修飾之后的鉑配合物,對TopoⅡ抑制活性明顯提高。鉑配合物是通過穩定拓撲異構酶-DNA二元復合物來達到抑制效果的,是典型的TopoⅡ毒劑。

圖11 鉑配合物拓撲異構酶抑制劑結構式
2.3.2 釕類配合物作為拓撲異構酶抑制劑
釕多吡啶配合物具有既為剛性又帶手性的八面體構型,水溶性比較好,熱力學性質穩定,不易發生配體取代,易于在近生理條件下開展研究,光化學、光物理信息豐富,毒性低,細胞膜透性較好。1999年,A.K.Kondapi報道配合物[RuCl2(C6H6)(dmso)]可以作為TopoⅡ毒劑,用于抑制腫瘤增殖[26]。而后,他們將DMSO換成嘧啶及嘧啶類衍生物,發現它們具有更好的TopoⅡ毒性,并對乳腺癌和結腸癌表現出更高的細胞毒性[27]。此后,關于以Topo為靶點的釕配合物的報道并不多。我們課題組長期致力于釕配合物抗腫瘤活性的研究。近年來,我們設計合成了一系列具有剛性平面配體的釕多吡啶配合物,并對其抗腫瘤作用機理進行了系統的研究。2007年,我們首次報道了具有TopoⅡ抑制效果的一對手性配合物 Λ-[Ru(bpy)2(uip)]2+和 Δ-[Ru(bpy)2(uip)]2+(圖12a)[28],結果顯示,它們對 TopoⅡ具有很強的抑制活性,IC50分別為3μmol/L與5μmol/L。通過抑制機理試驗,我們發現這一對手性配合物為TopoⅡ毒劑,可以穩定拓撲異構酶-DNA斷裂復合物,并且與傳統的TopoⅡ毒劑依托泊苷、玫瑰樹堿一樣,它們可以直接作用于拓撲異構酶。體外毒性結果顯示,配合物對人白血病細胞株、人肝癌細胞株、人宮頸癌細胞株、人鼻咽癌細胞株也表現了良好的抗癌活性。通過引入鳥嘌呤結構,合成了具有特異識別G-C堿基的釕(Ⅱ)多吡啶配合物[Ru(bpy)2(appo)]2+。抑酶活性測定證實該配合物表現出很高的TopoⅡ抑制能力(IC50<1μmol/L)[29]。進一步研究發現,配合物對拓撲異構酶的抑制活性與釕配合物輔助配體有一定關系[30]。最近,我們又相繼報道了4個具有良好TopoⅠ/Ⅱ雙抑制活性的釕多吡啶配合物[Ru(bpy)2(bfipH)]2+、[Ru(phen)2(bfipH)]2+(圖12b)、Λ/Δ-[Ru(bpy)2(ipad)]2+(圖12c)[31-32],該類配合物對 TopoⅠ/Ⅱ均具有良好的抑制活性,是TopoⅠ/Ⅱ的雙重毒劑。細胞毒性實驗顯示,配合物對人宮頸癌細胞株、人肝癌細胞株、人乳腺癌細胞株等有較好的抑制活性。通過細胞實時分析、單細胞凝膠電泳、AO/EB雙染實驗和流式細胞術等實驗,證明配合物可以造成細胞DNA損傷,進而導致細胞凋亡。目前釕類配合物作為拓撲異構酶抑制劑的抗腫瘤活性研究僅限于體外研究階段,需要更進一步對其作用機理等進行詳細的研究,以確定其作為抗腫瘤藥物的臨床可能性。

圖12 釕配合物拓撲異構酶抑制劑結構式
2.3.3 其他金屬配合物作為拓撲異構酶抑制劑
除了鉑、釕類金屬配合物以外,常見的已用于抗腫瘤藥物研究的還有金、鎘、鈷、鎳等金屬配合物,但是關于它們作為拓撲異構酶的抑制劑的報道則很少。
支志明課題組最近首次報道了4個單核與雙核金配合物,配合物包含一個三聯吡啶配體,與金原子形成共平面。研究結果顯示,該類配合物是TopoⅠ抑制劑,在小鼠體內顯示了很好的腫瘤抑制活性[33]。之后,A.Desideri課題組詳細研究了金配合物的拓撲異構酶抑制機理。研究發現,金配合物與喜樹堿類抑制機理完全不同,它是通過直接抑制拓撲異構酶的活性來抑制整個催化過程[34]。
此外,S.K.Singh等發現一系列有水溶性的芳烴金屬銠配合物有很好的 TopoⅡ抑制作用[35],D.Jayaraju等研究了一系列鈷水楊醛肟類配合物,發現它們是一類很好的DNA插入試劑和TopoⅡ毒劑,能夠誘導癌細胞凋亡[36]。近年來,又陸續有一些Cd2+、Ni2+[37-38]金屬配合物作為TopoⅡ抑制劑的報道。目前,金屬配合物抑制DNA拓撲異構酶的研究還處于起步階段,有關配合物結構、DNA作用方式及拓撲異構酶抑制功能間相互關系等方面的研究工作都急待開展。
3 結語
拓撲異構酶在DNA的復制、轉錄、翻譯、重組和修復中起著十分重要的作用,現已成為抗腫瘤治療的一種新靶點。從20世紀70年代發現DNA拓撲異構酶晶體結構至今,經過幾十年的努力,人們已經研制出一系列的拓撲異構酶抑制劑作為有效的抗腫瘤藥物應用于臨床,并對其作用機理做了大量研究。但是抑制劑殺傷癌細胞的機理十分復雜,癌細胞的最終死亡很可能是多種調控蛋白復制叉協同作用的綜合結果,而抑制所完成的僅是全部過程中的一個必要起始步驟。人們對這一系列的過程仍需進行更深入的研究。另外,作為拓撲異構酶抑制劑,金屬配合物有其獨特的優勢,雖然迄今這方面的研究還很少,但有望會在不久的將來成為一個研究熱點。
[1]Wang JC.Annu Rev Biochem,1996,65:635
[2]Champoux JJ.Annu Rev Biochem,2001,70:369
[3]Wang JC.Ann Rev Biochem,1985,54:665
[4]Li T K,Liu L F.Annu Rev Pharmacol Toxicol,2001,41:53
[5]Pommier Y.Chem Rev,2009,109:2894
[6]Christiansen K,Westerjaard O C.J Biol Chem,1994,269:721
[7]Austin C A,Marsh K L.BioEssays,1998,20:215
[8]Fortune J M,Osheroff N.Prog Nucleic Acid Res Mol Biol,2000,64:53
[9]張乃哲,江平.實用心腦肺血管病雜志,2002,10:121
[10]Larsen A K,Escargueil A E,Skladanowski A.Pharmacolo Ther,2003,99:167
[11]Wall M E,Wani M C.Cancer Res,1995,55:753
[12]Kejun C,Nicolas J R,Brian M E,et al.J Am Chem Soc,2005,127:838
[13]Song Y,Shao Z,Dexheimer T S,et al.J Med Chem,2010,53:1979
[14]Siu F M,Che C M.J Am Chem Soc,2008,130:17928
[15]Burden D A,Osheroff N.Biochim Biophys Acta,1998,1400:139
[16]Larsen A K,Escargueil A E,Skladanowski A.Pharmacol Ther,2003,99:167
[17]Goodell JR,Ougolkov A V,Hiasa H,et al.J Med Chem,2008,51:179
[18]Mistry A R,Felix C A,Whitmarsh R,et al.N Engl J Med,2005,352:1529
[19]Riou JF,Fosse P,Nguyen CH,et al.CancerRes,1993,53:5987
[20]Salerno S,Taliani S,Simorini F.Curr Med Chem,2010,17:4270
[21]Gijn R,Beijnen JH,Bult A,et al.J Oncol Pharm Pract,2000,6:92
[22]Van Waardenburg R C,De Jong L A,Van Eijndhoven M A,et al.J Biol Chem,2004,279:54502
[23]Lo Y C,Ko T P,Su W C,et al.J Inorg Biochem,2009,103:1082
[24]Wang P,Leung C H,Ma D L,et al.Chem Asian J,2010,5:2271
[25]Liu J,Leung C H,Chow A L,et al.Chem Commun,2011,47:719
[26]Gopal Vashisht Y N,Jayaraju D,Kondapi A K.Biochemistry,1999,38:4382
[27]Gopal Vashisht Y N,Konuru N,Kondapi A K.Arch Biochem Biophys,2002,401:53
[28]Gao F,Chao H,Wang J Q,et al.J Biol Inorg Chem,2007,12:1015
[29]Gao F,Chao H,Zhou F,et al.J Inorg Biochem,2008,102:1050
[30]Chen X,Gao F,Zhou Z X,et al.J Inorg Biochem,2010,104:576
[31]Du K J,Wang JQ,Kou J F,et al.Eur J Med Chem,2011,46:1056
[32]Kou J F,Qian C,Wang JQ,et al.J Biol Inorg Chem,2012,17:81
[33]Yan J J,Chow A L F,Leung C H,et al.Chem Commun,2010,46:3893
[34]Castelli S,Vassallo O,Katkar P,et al.Arch Biochem Biophys,2011,516:108
[35]Singh SK,Joshi S,Singh A R.Inorg Chem,2007,46:10869
[36]Jayaraju D,Vashisht Y N,Anand K.Arch Biochem Biophys,1999,369:68
[37]Wu Xing,Jack C,Brian B.J Inorg Biochem,2011,105:833
[38]Prabhakaran R,Sivasamy R,Angayarkanni J,et al.Inorg Chim Acta,2011,105:833
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
