水氯失衡對連續重整催化劑性能的影響
2013-07-19 02:52:40劉淑敏馬愛增
石油煉制與化工 2013年2期
關鍵詞:催化劑
劉淑敏,馬愛增
(中國石化石油化工科學研究院,北京100083)
石腦油催化重整過程是在催化劑的作用下,將石腦油轉化為芳烴、高辛烷值汽油組分和氫氣的過程,該過程主要包含六元環烷烴脫氫、五元環烷烴脫氫異構化、鏈烷烴脫氫環化、鏈烷烴異構化、鏈烷烴氫解、加氫裂化和結焦等反應。實現這些反應要求催化劑既具有金屬活性中心,又具有酸活性中心,這兩種活性中心分別提供金屬加氫-脫氫功能和酸性異構化功能。金屬功能主要由金屬Pt提供,酸性功能由氧化鋁提供。氧化鋁本身只有很弱的酸性,不能滿足重整反應的要求。鹵素的引入可以提高氧化鋁的酸性,并且氧化鋁的酸性隨著氯含量的增加而線性增加[1],因此重整催化劑的酸性可以通過氯含量進行調控。
重整催化劑上的氯在含水和高溫環境容易流失,并且隨著水含量和溫度的升高,流失速率加快。然而催化重整的反應和燒焦過程均是在高溫和含水氣氛中進行,因此催化劑上的氯含量會不斷降低,為了保證催化劑具有足夠的酸性,就必須不斷注氯,補充流失的氯,保持催化劑具有足夠的氯含量。因此,催化重整過程中水氯平衡的控制非常重要[2-7]。本課題針對某公司600kt/a連續重整工業裝置水氯失衡進行跟蹤,研究水氯失衡對重整催化劑性能的影響。
1 實 驗
1.1 催化劑金屬分散度測定
采用Micromeritics公司生產的Autochem 2920型化學吸附儀對催化劑樣品的金屬分散度進行測定,所用方法為氫氧滴定法。將催化劑樣品研碎后取20~40目的顆粒,首先用氬氣和氫氣的混合氣在500℃對催化劑進行還原,然后降至室溫進行氧氣吸附,之后分別在35℃和170℃進行氫氣脈沖滴定,直到脈沖峰的峰面積不再變化。氫氣滴定過程中的化學計量關系見文獻[8],利用Autochem 2920附帶的軟件進行數據處理,得到催化劑的分散度。
1.2 工業應用連續重整催化劑在實驗室條件下的氯化還原
采集工業裝置上正在運轉的連續重整催化劑,在實驗室進行氯化更新,實驗條件為:常壓,490~510℃,空氣介質,控制合適的水氯摩爾比,時間6~8h。氯化更新后的催化劑用高純氫氣將金屬組元從氧化態還原成金屬態,還原溫度450~500℃,時間4~6h。
1.3 催化劑的反應性能評價
催化劑反應性能評價在自建的帶循環壓縮機的中型重整評價裝置上進行,原料油性質見表1。反應條件為:壓力0.69MPa,體積空速2h-1,氫油體積比800,反應器入口溫度530℃。

表1 中型反應裝置原料油的性質
1.4 比表面積的測定
催化劑的比表面積采用低溫氮吸附法測定,所用儀器為Micromeritics公司生產的ASAP2400靜態氮吸附儀。樣品在1.33Pa、300℃下抽真空脫氣4h,以氮氣為吸附質,在77.4K下等溫吸附、脫附,測定脫附等溫線,用BET公式計算比表面積。
1.5 積炭量的測定
催化劑上的積炭量在高頻燃燒紅外吸收硫炭儀上測定。所用儀器為美國LECO公司生產的CS-344型紅外硫炭測定儀。先將樣品在110~120℃下烘2h,冷卻至室溫,然后將裝有樣品的坩堝放在電子天平上,加入助熔劑,在純度高于99.5%的氧氣流中燃燒,通過紅外感應,測定積炭量。
1.6 氯含量的測定
催化劑的氯含量采用氯離子選擇性電極法分析。所用儀器為江蘇太倉生產的PXJ-10型數字式離子計數器。取研細的樣品0.06~0.10g,用氫氧化鈉溶液進行抽提,加入一定量的硝酸溶液調節pH值為7~9。將儀器的電極插入配置好的樣品中,通過與空白溶液的對比測定氯的含量。
1.7 X射線衍射(XRD)
XRD分析采用Philips公司生產的X-P’ert型X射線衍射儀,Cu靶,Kα輻射,Ni濾波,λ=0.154 056nm;固體探測器,管電壓40kV,管電流40mA,掃描范圍5°~70°。
1.8 TEM透射電子顯微鏡(TEM)
TEM分析采用荷蘭FEI公司生產的TecnaiG2F20S-TWIN型透射電鏡,加速電壓200kV。分析測試前將樣品研細,放入無水乙醇中超聲分散后,滴加到擔載碳膜的銅網上觀察樣品。
2 結果與討論
2.1 連續重整工業裝置的進料組成及操作條件
某公司600kt/a連續重整裝置采用美國UOP公司開發的超低壓(0.35MPa)重整反應工藝,催化劑連續再生部分采用CycleMax技術,催化劑采用中國石化石油化工科學研究院研發的PS-Ⅴ催化劑。重整進料的組成見表2,重整反應部分和再生部分的主要操作條件見表3和表4。
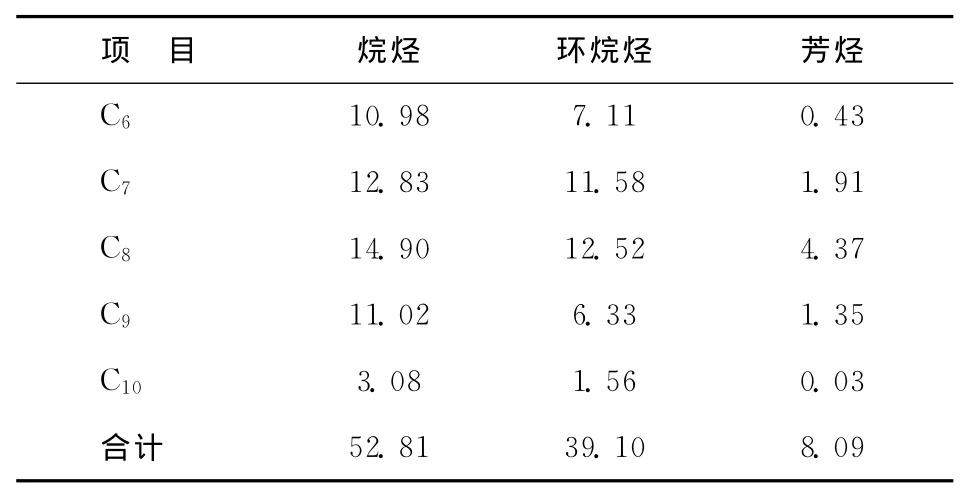
表2 重整進料的組成 w,%
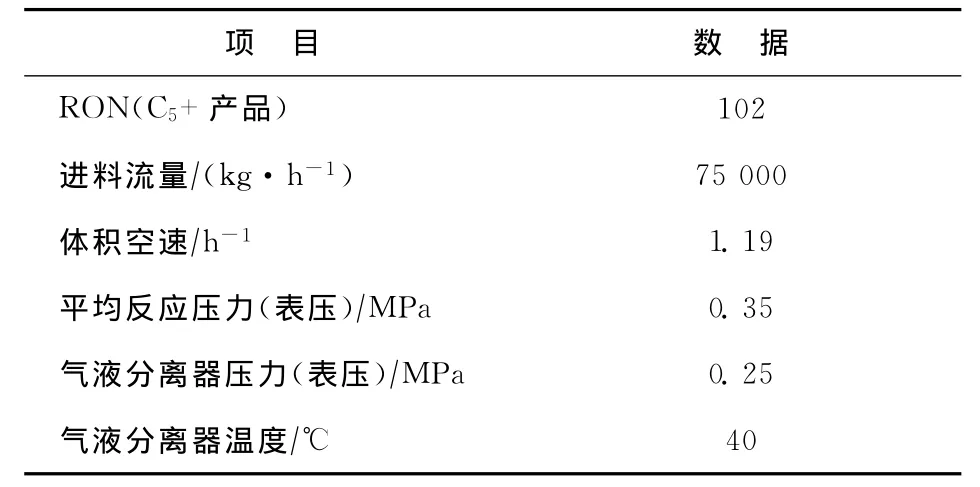
表3 反應部分的主要操作條件
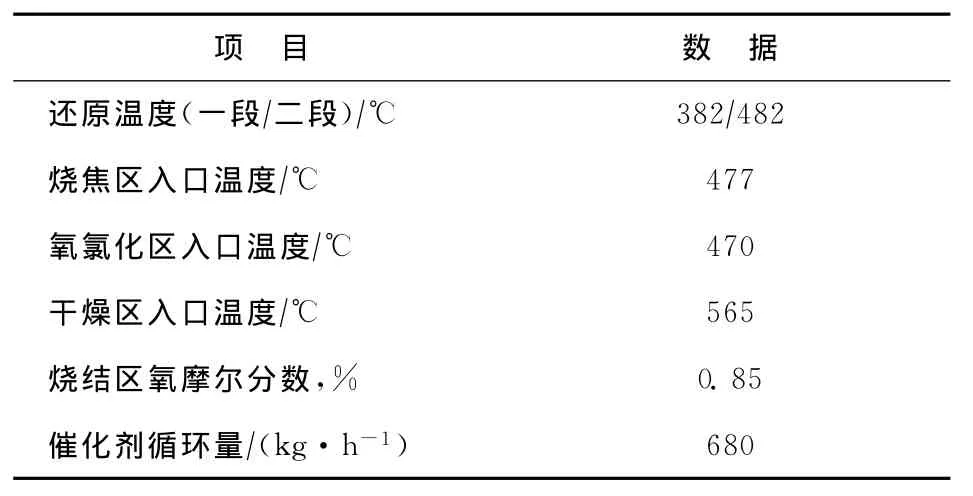
表4 再生部分的主要操作條件
2.2 工業運轉催化劑物化性質變化
600kt/a連續重整裝置于2002年7月開工,對催化劑的物化性質變化情況進行了跟蹤分析,氯含量及主要雜質含量變化見表5,比表面積的變化見圖1。
從表5可以看出,隨著運轉時間延長,催化劑氯含量逐漸下降,運轉1 967天后,催化劑的氯質量分數由開工初期的1.20%下降到0.88%。對于PS-Ⅴ催化劑,適宜的氯質量分數為1.0%~1.2%;運轉1 482天后,催化劑的氯質量分數就已經低于1.0%的下限,在1 482~2 147天期間,催化劑的氯質量分數一直低于1.0%。
對運轉催化劑氯含量逐漸降低的原因進行分析,發現該裝置水氯調節失衡,重整進料中水含量較高,同時氮質量分數超標(大于0.5μg/g),有銨鹽形成并造成了嚴重腐蝕與堵塞。為了避免銨鹽形成,對該裝置采取了降低注氯量的操作方式,由此導致催化劑氯含量逐漸下降。
從表5還可以看出,催化劑的Fe含量在開工初期增加較快,運轉1 117天后,Fe質量分數由開工初期的150μg/g增加到1 800μg/g;運轉1 967天后,Fe質量分數增加到2 400μg/g,隨后基本保持在這一水平。催化劑的Si,Na,S含量在不同時期略有變化,但變化幅度不大。
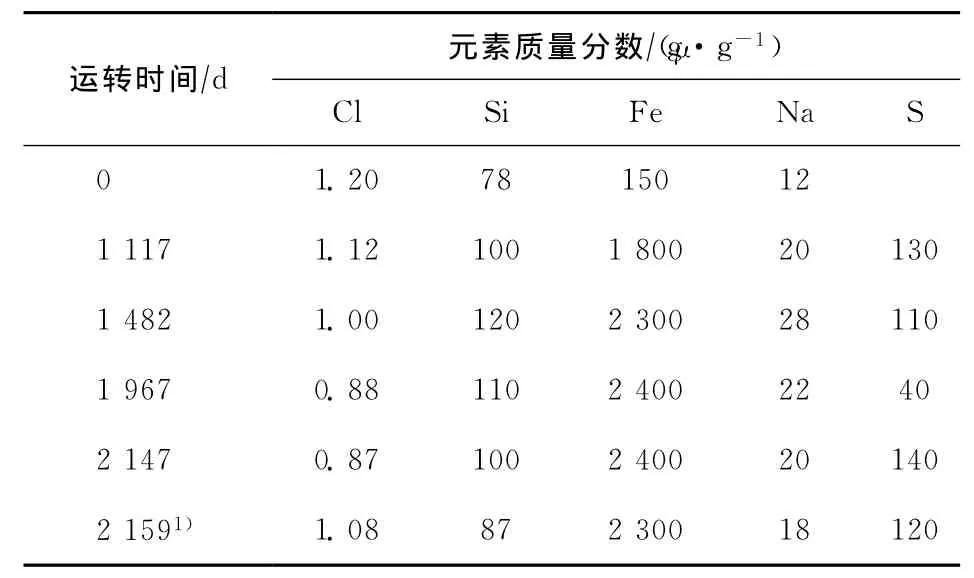
表5 催化劑氯含量及主要雜質含量變化情況
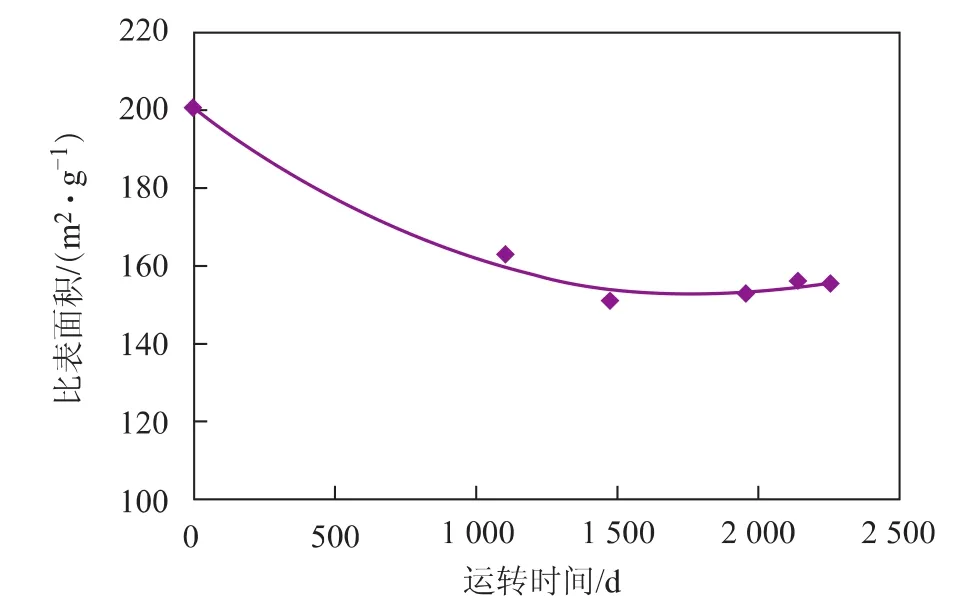
圖1 催化劑比表面積隨運轉時間的變化
由圖1可見,新鮮催化劑的比表面積為200 m2/g,在運轉初期,催化劑比表面積下降較快,運轉至1 500天左右,催化劑比表面積降至最低,約為150m2/g;隨后催化劑比表面積趨于穩定,并略有增加,主要是由于補充的新催化劑比表面積較高引起的。
2.3 催化劑催化性能變化
為了考察催化劑性能隨運轉時間的變化,選取新鮮催化劑、運轉1 482天和1 967天后的催化劑在中型實驗裝置上進行催化性能評價,結果見表6。從表6可以看出:與新鮮催化劑相比,采用運轉1 482天的催化劑時液體收率增加1.29百分點,C5+產物中芳烴含量下降1.27百分點,芳烴產率下降0.1百分點,C5+產物的RON下降0.5個單位,積炭量下降0.12百分點,表明催化劑的活性略有下降,這主要是由于催化劑Fe雜質含量增加引起,符合連續重整催化劑工業運轉的一般規律;運轉1 967天的催化劑表現出異常的催化性能,液體收率增加0.47百分點,C5+產物中芳烴含量下降12.89百分點,芳烴產率下降10.59百分點,C5+產物的RON下降4.23個單位,積炭量增加0.77百分點,表明催化劑的活性、選擇性均大幅度下降,而該催化劑的Fe質量分數為2 400μg/g(見表5),僅比運轉1 482天的催化劑高100μg/g,不符合連續重整催化劑工業運轉的一般規律。
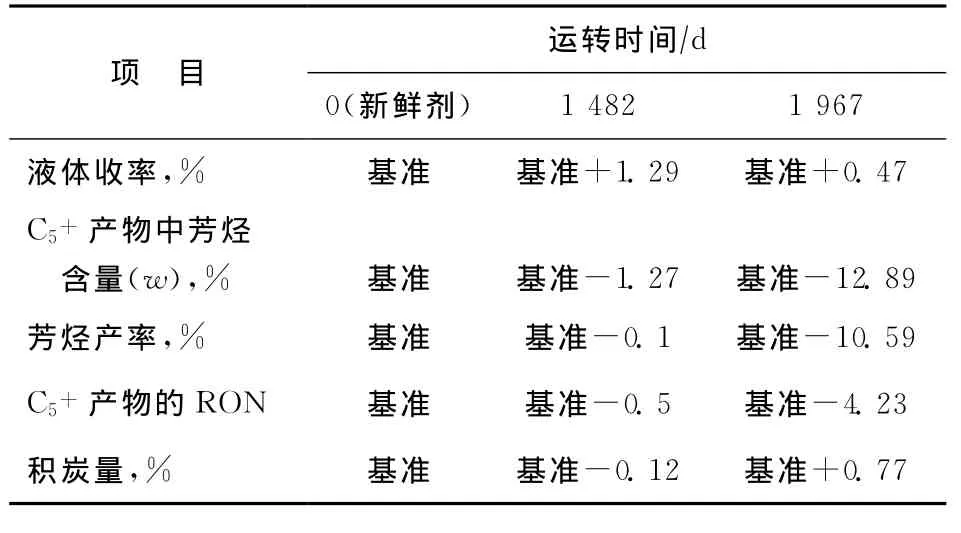
表6 不同運轉時間的催化劑的催化性能
運轉1 967天的催化劑與新鮮催化劑的催化性能對比見圖2~圖4。由圖2可見,采用運轉1 967天的催化劑時初始芳烴產率較低,意味著初活性較低,并且隨著反應的進行,芳烴產率下降較快,說明催化劑活性下降較快。由圖3可見,采用運轉1 967天的催化劑時初始液體收率較低,但隨著反應的進行,液體收率逐漸增大并超越新鮮催化劑,參照芳烴產率的變化可知,這主要是催化劑活性降低、轉化率下降引起的。由圖4可見,采用運轉1 967天的催化劑時液體收率下降幅度較大,并且在相同芳烴含量時,液體收率較低,表明催化劑的選擇性降低。
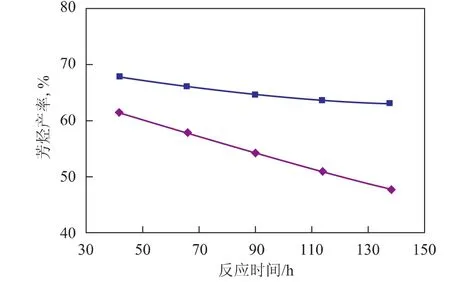
圖2 芳烴產率隨反應時間的變化
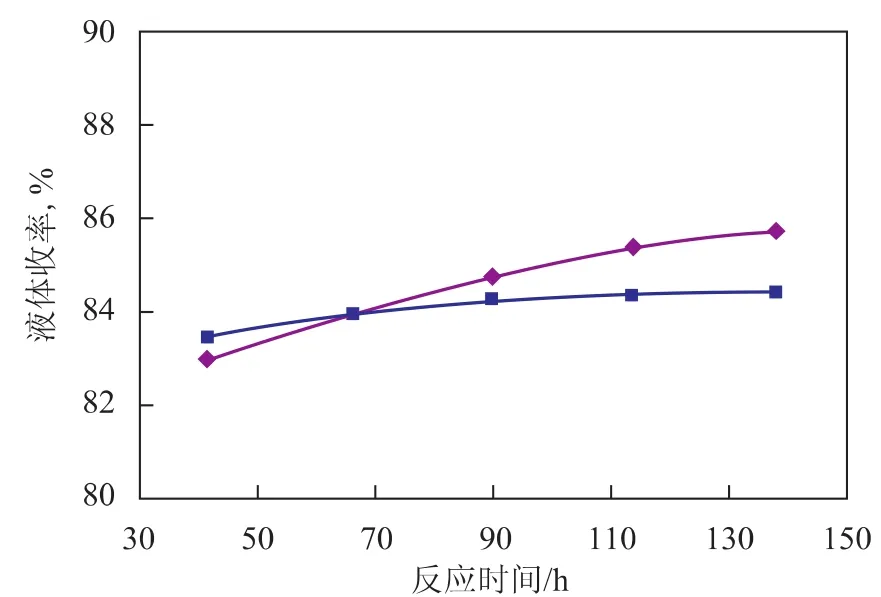
圖3 液體收率隨反應時間的變化
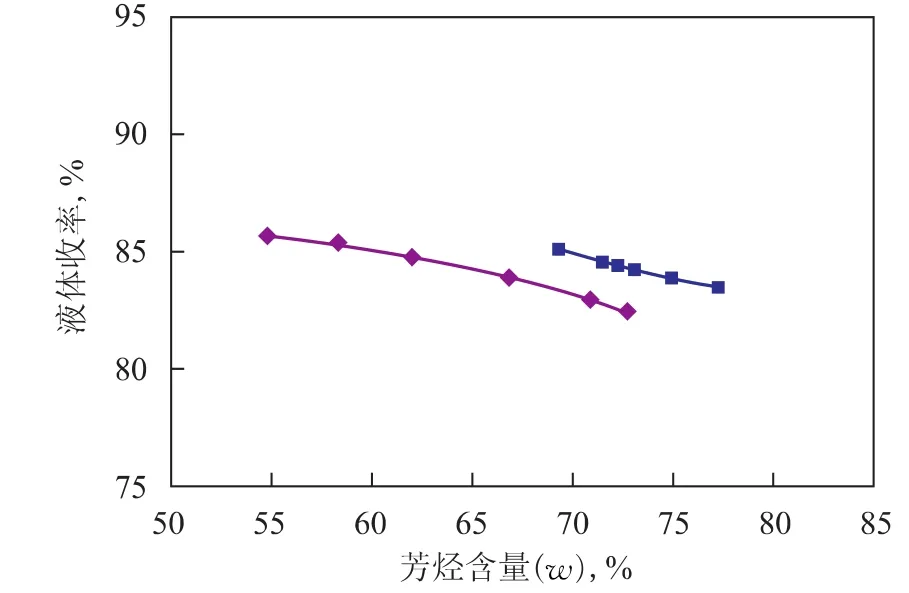
圖4 液體收率與芳烴含量的關系
2.4 催化劑催化性能變差的原因分析
為了考察運轉催化劑失活的原因,對運轉催化劑及新鮮催化劑進行了物化表征。不同運轉時間催化劑的鉑分散度見表7。運轉1 967天的催化劑的XRD圖譜見圖5。在實驗室對運轉1 967天的催化劑進行氧氯化和還原,氧氯化溫度510℃,時間8h;還原溫度500℃,時間6h。不同部位運轉1 967天的催化劑在氧氯化和還原處理前后的TEM照片見圖6。
由表7可見,新鮮催化劑的鉑分散性較好,n(H)/n(Pt)為1.00;運轉1 482天催化劑的n(H)/n(Pt)為 0.98;運轉 1 967 天催化劑的n(H)/n(Pt)大幅度下降,僅為0.44。由圖5可見,運轉1 967天的催化劑在2θ為40°附近出現了1個新衍射峰,此衍射峰不是氧化鋁的衍射峰,而是積聚的Pt晶粒的衍射峰[9],進一步說明運轉1 967天的催化劑中鉑發生了積聚。由圖6(a)~(c)可見,運轉1 967天的催化劑的TEM照片中出現大晶粒的Pt,最大晶粒直徑達到90nm,進一步證明運轉催化劑中的Pt發生了嚴重積聚。由此可以推測,Pt的嚴重積聚導致Pt分散度大幅度下降是引起運轉1 967天的催化劑催化性能大幅度下降的主要原因。

表7 不同運轉時間的催化劑的鉑分散度
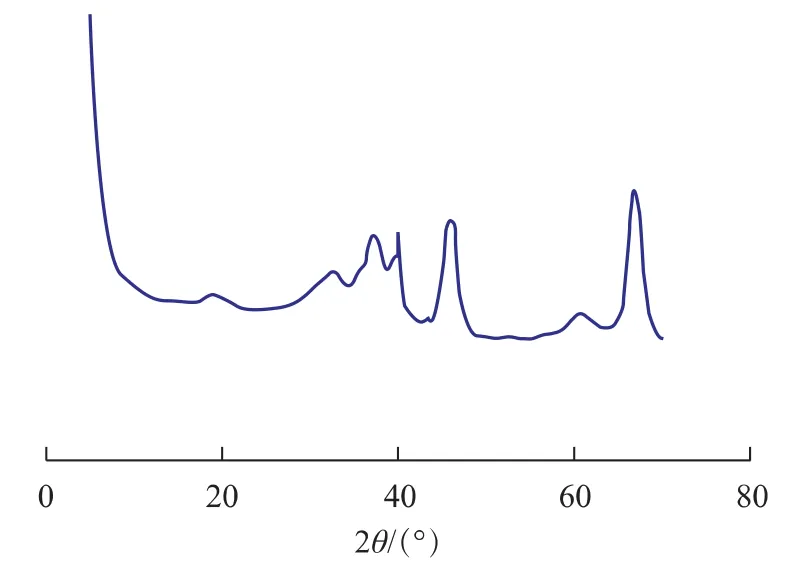
圖5 運轉1 967天催化劑的XRD圖譜
2.5 運轉催化劑中Pt的再分散
一般認為,氧氯化和還原過程是使積聚的Pt晶粒再分散的主要過程。在氧氯化過程中,引入的氯離子與Pt晶粒反應生成[PtOxCly]s表面絡合物[10],它可以沿催化劑表面遷移,或由氣相傳遞到載體表面高能位上,形成新的晶粒。在還原過程中,經氫氣還原后,催化劑中氧化態的Pt得到還原。因此,催化劑上的氯一方面可以增強氧化鋁的酸性,另外一個主要的貢獻是使催化劑的Pt處于高分散狀態,催化劑上氯含量的降低,可以造成催化劑的Pt積聚,因此對于每一種商用催化劑,都有一個推薦的氯含量范圍。
由圖6(d)~(f)可見,運轉1 967天的催化劑在氧氯化和還原后,其TEM照片中依然觀察到積聚的Pt,且晶粒沒有明顯減小,說明嚴重積聚的Pt晶粒在常規氧氯化和還原條件下很難再分散。

圖6 運轉1 967天的催化劑在氧氯化和還原處理前后的TEM照片
在發現催化劑氯含量偏低后,對該套裝置進行了水氯操作的調節,增加了注氯量。由表5可見,運轉2 159天時催化劑的氯質量分數已經提高到1.08%。由表7可見,盡管氯含量提高,但運轉2 159天催化劑的n(H)/n(Pt)僅為0.45,沒有明顯變化,進一步說明盡管氧氯化和還原可以使積聚的Pt晶粒得到再分散,但是在常規氧氯化和還原條件下,特別是在連續重整裝置的操作條件下,嚴重積聚的Pt晶粒的再分散速率非常慢,很難使運轉催化劑恢復到新鮮催化劑的Pt分散水平,從而導致催化劑性能大幅度下降。
3 結 論
(1)催化重整工業運轉裝置長時間的水氯失衡,不僅可導致催化劑氯含量降低,還使催化劑中的Pt發生嚴重積聚,最大Pt晶粒直徑達到90nm,并在2θ為40°附近出現了衍射峰。
(2)催化劑中Pt的嚴重積聚,導致催化劑活性和選擇性大幅度下降。催化劑運轉1 967天時,C5+產物中芳烴含量下降12.89百分點,芳烴產率下降10.59百分點,C5+產物的RON下降4.23個單位,積炭量增加0.77百分點。
(3)盡管氧氯化和還原可以使積聚的Pt晶粒得到再分散,但是在常規氧氯化和還原條件下,特別是在連續重整裝置的操作條件下,嚴重積聚的Pt晶粒的再分散速率非常慢,很難使運轉催化劑恢復到新鮮催化劑的Pt分散水平。
[1]Tanaka M,Ogasawara S.Infrared studies of the adsorption and the catalysis of hydrogen chloride on alumina and on silica[J].J Catal,1970,16:157-163
[2]任蔚.調整重整水氯平衡提高穩定汽油質量[J].化學工程與裝備,2008(8):38-40
[3]楊利平.淺析重整催化劑水氯平衡的調整[J].化學工程與裝備,2011(12):65-68
[4]王安川.水-氯平衡對雙金屬重整催化劑性能的影響[J].石油煉制,1990,21(11):29-34
[5]孫策,張文娟,孫雋.水氯平衡調整在催化劑上的工業應用[J].煉油技術與工程,2009,39(11):20-23
[6]莫治兵,張明耀,費長林.重整催化劑水氯平衡控制[J].石化技術與應用,2008,26(4):365-369
[7]戴永川,戴承遠.用循環氣組成指導重整裝置的水氯調整[J].石油與天然氣化工,1998,27(4):222-224
[8]George J A,Abdullah M A.Catalytic naphtha reforming[M].2nd Edition.New York:Marcel Dekker,2004:199-262
[9]劉辰,馬愛增.工業連續重整催化劑的Pt積聚與再分散研究[J].石油煉制與化工,2010,41(8):29-33
[10]徐澤輝.重整催化劑Pt晶粒的再分散[J].工業催化,1996(3):55-59
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
