達原滴丸質量標準研究*
2012-12-23 04:12:26畢麗萍
天津藥學 2012年3期
杜 平,曲 佳,畢麗萍
(1. 天津市傳染病醫院,天津 300192; 2.天津市藥品檢驗所,天津 300070)
達原滴丸處方源于明代吳又可《瘟疫論》中的達原飲,由檳榔、厚樸、草果、芍藥、知母、黃芩、甘草組成,為開達膜原、避穢化濁之要方,主治瘟疫或瘧疾邪伏膜原之證。現代多應用于病毒感染性發熱、濕熱發熱以及一些不明原因的高熱、盜汗、病毒性腦炎等[1]。達原滴丸是在達原飲方劑的基礎上研制出的中藥制劑,為了有效控制該制劑質量,本實驗采用薄層色譜法鑒別處方中的白芍、知母、檳榔、厚樸及甘草;采用高效液相色譜法測定處方中芍藥苷的含量。
1 儀器和試藥
1.1 儀器 SHIMADZU LC -2010AHT 高效液相色譜儀,儀器工作站:LC -Solution,色譜柱:phenomenex Luna C18(250 mm×4.6 mm,5 μm)色譜柱。
1.2 試藥 知母對照藥材(批號121070 -200804)、檳榔對照藥材(批號120915 -201011)、厚樸酚對照品(批號110729 - 200412)、和厚樸酚對照品(批號110730 - 200609)、甘草對照藥材(批號120904 -201016)、芍藥苷對照品(批號110736 -200933)均購自中國藥品生物制品檢定所;薄層層析用硅膠G 板(青島海洋化工廠),乙腈、甲醇為色譜純,水為去離子水,其他試劑均為分析純。
2 方法與結果
2.1 白芍薄層鑒別
2.1.1 供試品溶液制備 取本品20 丸,研碎,加乙醇20 ml,超聲處理30 min,濾過,濾液蒸干,殘渣加乙醇1 ml 使溶解,作為供試品溶液。
2.1.2 對照品溶液制備 取芍藥苷對照品,加乙醇制成每1 ml 含1 mg 的溶液,作為對照品溶液。
2.1.3 陰性對照溶液制備 按處方配比,取除白芍外的其他藥材,按工藝制成滴丸劑,再按“2.1.1”項下方法制得陰性對照溶液。
2.1.4 薄層條件及結果 照薄層色譜法(《中國藥典》2005 年版一部附錄ⅥB)試驗,吸取上述三種溶液各10 μl,分別點于同一硅膠G 薄層板上,以三氯甲烷-乙酸乙酯-甲醇-甲酸(40∶ 5∶ 10∶ 0.2)為展開劑,展開,取出,晾干,噴以5%香草醛硫酸溶液,加熱至斑點顯色清晰。供試品色譜中,在與對照品色譜相應的位置上,顯相同顏色的斑點,陰性對照溶液無干擾。結果見圖1。
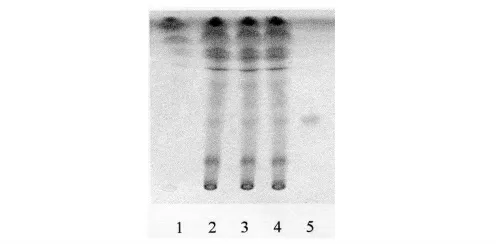
圖1 白芍TLC 色譜圖
2.2 知母薄層鑒別
2.2.1 供試品溶液制備 取本品20 丸,研碎,加乙醇20 ml,加熱回流40 min,濾過,取濾液10 ml,加鹽酸1 ml,加熱回流1 h 后濃縮至約5 ml,加水10 ml,用甲苯20 ml 振搖提取,提取液蒸干,殘渣加甲苯1 ml 使溶解,作為供試品溶液。
2.2.2 對照藥材溶液制備 取知母對照藥材2 g,同法制得對照藥材溶液。
2.2.3 陰性對照溶液制備 按處方配比,取除知母外的其他藥材,按工藝制成滴丸劑,再按“2.2.1”項下方法制得陰性對照溶液。
2.2.4 薄層條件及結果 照薄層色譜法(《中國藥典》2005 年版一部附錄ⅥB)試驗,吸取上述三種溶液各10 μl,分別點于同一硅膠G 薄層板上,以甲苯-丙酮(9∶ 1)為展開劑,展開,取出,晾干,噴以5%香草醛硫酸溶液,加熱至斑點顯色清晰。供試品色譜中,在與對照藥材色譜相應的位置上,顯相同顏色的斑點,陰性對照溶液無干擾。結果見圖2。
2.3 檳榔薄層鑒別
2.3.1 供試品溶液制備 取本品20 丸,研碎,加三氯甲烷20 ml 及濃氨試液3 ml ,超聲處理30 min,濾過,濾液加稀鹽酸10 ml、水20 ml,振搖,分取酸水層,加氨試液,調節pH 值至8 ~9 ,用三氯甲烷振搖提取2 次,每次10 ml,合并三氯甲烷提取液,蒸干,殘渣加甲醇0.5 ml 使溶解,作為供試品溶液。
2.3.2 對照藥材溶液制備 取檳榔對照藥材1 g,同法制成對照藥材溶液。
2.3.3 陰性對照溶液制備 按處方配比,取除檳榔外的其他藥材,按工藝制成滴丸劑,再按“2.3.1”項下方法制得陰性對照溶液。
2.3.4 薄層條件及結果 照薄層色譜法(《中國藥典》2005 年版一部附錄ⅥB)試驗,吸取上述三種溶液各10 μl,分別點于同一用1%氫氧化鈉溶液制備的硅膠G 薄層板上,以甲苯-三氯甲烷-甲醇(10∶ 4∶ 1)為展開劑,展開,取出,晾干,噴以碘化鉍鉀試液。供試品色譜中,在與對照藥材色譜相應的位置上,顯相同顏色的斑點,陰性對照溶液無干擾。結果見圖3。
2.4 厚樸薄層鑒別
2.4.1 供試品溶液制備 取本品20 丸,研碎,加甲醇20 ml,超聲處理30 min,濾過,濾液蒸干,殘渣加甲醇1 ml 使溶解,作為供試品溶液。
2.4.2 對照品溶液制備 取厚樸酚對照品、和厚樸酚對照品,加甲醇制成每1 ml 各含1 mg 的混合溶液,作為對照品溶液。
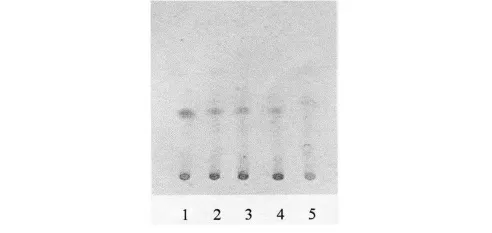
圖3 檳榔TLC 色譜圖
2.4.3 陰性對照溶液制備 按處方配比,取除厚樸外的其他藥材,按工藝制成滴丸劑,再按“2.4.1”項下方法制得陰性對照溶液。
2.4.4 薄層條件及結果 照薄層色譜法(《中國藥典》2005 年版一部附錄ⅥB)試驗,吸取上述三種溶液各10 μl,分別點于同一硅膠GF254薄層板上,以甲苯-甲醇(27∶ 1)為展開劑,展開,取出,晾干,置紫外光燈(254 nm)下檢視。供試品色譜中,在與對照品色譜相應的位置上,顯相同顏色的斑點,陰性對照無干擾。結果見圖4。
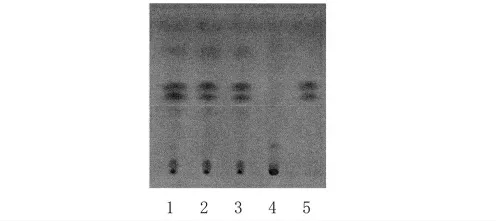
圖4 厚樸TLC 色譜圖
2.5 甘草薄層鑒別
2.5.1 供試品溶液制備 取本品20 丸,加乙醚40 ml,加熱回流1 h,濾過,棄去醚液,藥渣加甲醇30 ml,加熱回流1 h,濾過,濾液蒸干,殘渣加水40 ml 使溶解,用正丁醇提取3 次,每次20 ml,合并正丁醇液,用水洗滌3 次,棄去水液,正丁醇液蒸干,殘渣加甲醇5 ml 使溶解,作為供試品溶液。
2.5.2 對照藥材溶液制備 取甘草對照藥材1 g,同法制得對照藥材溶液。
2.5.3 陰性對照溶液制備 按處方配比,取除甘草外的其他藥材,按工藝制成滴丸劑,再按“2.5.1”項下方法制得陰性對照溶液。
2.5.4 薄層條件及結果 照薄層色譜法(《中國藥典》2005 年版一部附錄VI B)試驗,吸取上述三種溶液各10 μl,分別點于同一硅膠G 薄層板上,以乙酸乙酯-甲酸-冰醋酸-水(15∶ 1∶ 1∶ 2)為展開劑,展開,取出,晾干,噴以10%硫酸乙醇溶液,加熱至斑點顯色清晰,分別置日光及紫外光燈(365 nm)下檢視。供試品色譜中,在與對照藥材色譜相應的位置上,分別顯相同顏色的斑點(見圖5A)或熒光斑點(見圖5B)。陰性對照溶液無干擾。
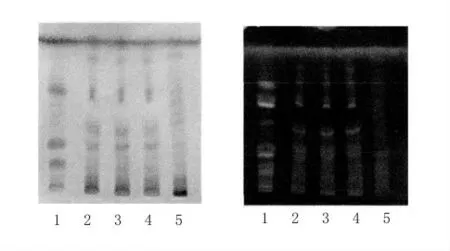
圖5 甘草TLC 色譜圖
2.6 白芍含量測定
2.6.1 色譜條件 參照《中國藥典》2005 年版一部制定[2]。以十八烷基硅烷鍵合硅膠為填充劑;以乙腈-磷酸鹽緩沖液[0.067 mol/L磷酸氫二鈉-0.067 mol/L磷酸二氫鉀(5∶ 1)配制成pH 7.4 緩沖液](15∶ 85)為流動相;檢測波長為230 nm。理論板數按芍藥苷峰計算應不低于3 000。
2.6.2 溶液制備
2.6.2.1 對照品溶液制備 取芍藥苷對照品適量,精密稱定,加甲醇制成每1 ml 含20 μg 的溶液,即得。
2.6.2.2 供試品溶液制備 取本品(批號D200601)適量,研細,取約1 g,精密稱定,置具塞錐形瓶中,精密加入稀乙醇50 ml,密塞,稱定重量,超聲處理45 min,放冷,再稱定重量,用稀乙醇補足減失的重量,搖勻,濾過,取續濾液,即得。
2.6.2.3 陰性對照溶液制備 按處方配制諸藥,取除白芍外的其他藥味,制成滴丸劑,再按“2.6.2.2”項下方法制成白芍陰性對照溶液。試驗結果表明陰性對照溶液無干擾。依“2.6.1”項下色譜條件,吸取對照品、供試品和陰性對照溶液注入色譜儀,記錄色譜圖,見圖1。
2.6.3 線性關系考查 取芍藥苷對照品適量,精密稱定,加甲醇制成每1 ml 含0.042 17 mg 的溶液,分別精密吸取1、2、5、7 和10 ml 置10 ml 量瓶中,加甲醇至刻度,搖勻,即得一系列對照品稀釋液。精密吸取上述系列對照品稀釋液5 μl,注入高效液相色譜儀,測定峰面積,以對照品溶液濃度(mg/ ml)為橫坐標,峰面積為縱坐標,計算回歸方程及相關系數。結果回歸方程為Y =2.448 2 ×106X + 6.330 7 ×102(r = 0.999 9)芍藥苷進樣量在0.021 1 ~0.210 8 μg 之間,線性關系良好。
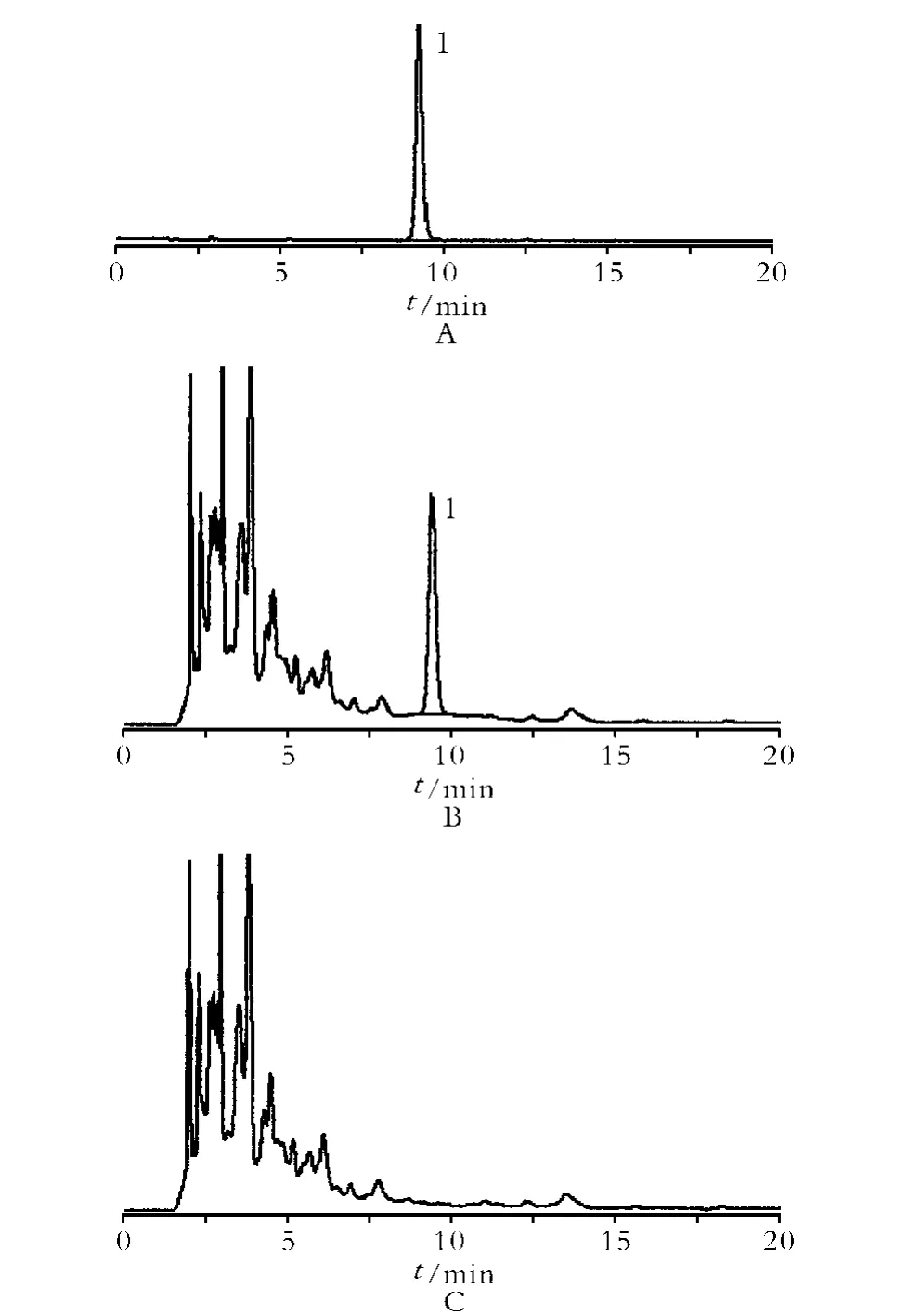
圖6 對照品(A)供試品(B)陰性對照(C)HPLC 色譜圖
2.6.4 重復性試驗 取本品(批號D200601)6 份,按“2.6.2.2”項下方法操作,按“2.6.1”項下色譜條件分析,樣品中芍藥苷平均含量為1.095 9 mg/g,RSD 為1.7%。
2.6.5 精密度試驗 取重復性試驗1 號樣品,按“2.6.1”項下色譜條件分析,連續進樣6 次,測得供試品溶液中芍藥苷峰面積值的RSD 為0.4%。
2.6.6 穩定性試驗 取重復性試驗1 號樣品,分別在0、4、8、12、16 和24 h 按“2.6.1”項下色譜條件分析,各進樣1 次,測得供試品溶液中芍藥苷峰面積值的RSD為0.5%,結果表明,供試品溶液在24 h 內穩定。
2.6.7 加樣回收率試驗 取本品(批號D200601),研細,稱取0.5 g,精密稱定,共6 份,精密加入對照品溶液50 ml(0.011 6 mg/ml),再按“2.6.2.2”項下供試品溶液制備方法操作,按“2.6.1”項下色譜條件分析,平均加樣回收率為102.05%,RSD 為1.5%。結果見表1。
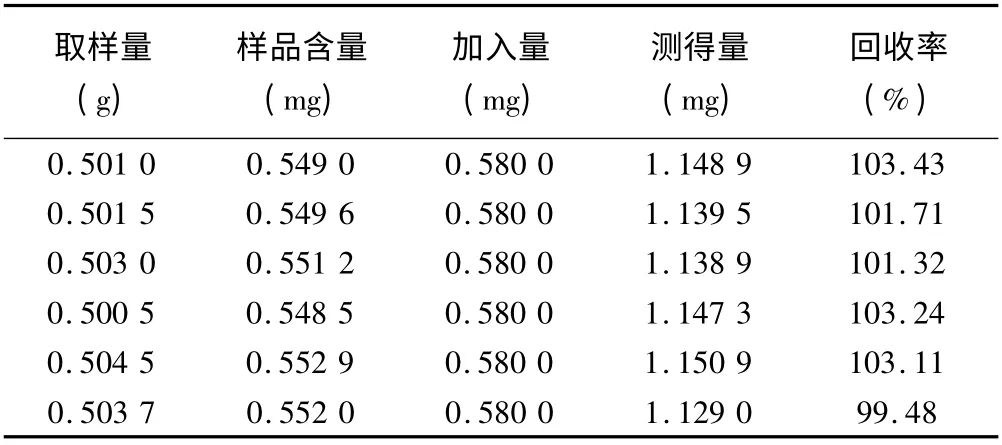
表1 加樣回收試驗(n=6)
2.6.8 樣品測定 分別取3 批樣品,按“2.6.2.2”項下供試品溶液制備方法操作,按“2.6.1”項下色譜條件分析。結果見表2。

表2 樣品含量測定
3 討論
3.1 波長的選擇 通過對芍藥苷對照品溶液在200 ~400 nm 處進行紫外光譜掃描檢測顯示,芍藥苷在230 nm 處有最大吸收,與文獻[2]采用的波長相同。
3.2 提取方法的選擇
3.2.1 提取方式的確定 分別考查了超聲處理與加熱回流的方式,結果顯示兩種提取方法基本一致,故采用相對簡便的超聲處理方式。
3.2.2 提取溶劑的選擇 分別采用甲醇、乙醇及稀乙醇為提取溶劑,結果顯示,以稀乙醇作為提取溶劑時,芍藥苷含量最高。
3.2.3 提取時間的選擇 分別考查了超聲處理30、45及60 min,結果顯示,提取45 min 與60 min 測定結果基本一致,故超聲處理時間選用45 min。
1 廖茂梁,張鐵軍,高文遠,等.大孔吸附樹脂純化達原滴丸處方提取液的工藝研究. 中草藥,2008,39(1):54
2 中國藥典.一部.2005:635
