合成苯并咪唑類化合物的工藝改進
2012-11-21 06:02:42白翠冰李軍艦張有明魏太保
合成化學 2012年3期
白翠冰, 李軍艦, 張 鵬, 張有明, 魏太保
(西北師范大學 化學化工學院 甘肅省高分子材料重點實驗室,甘肅 蘭州 730070)
苯并咪唑是一類具有多種生物生理活性的雜環化合物,由于其具有優良的結構特性、反應活性、生理藥物功效及配位性能,應用極為廣泛[1~3]。苯并咪唑衍生物不僅可以用作室溫離子液體系列的綠色溶劑,而且作為新型材料在超分子化學領域展示了良好的應用前景[4]。其中,2-烷基苯并咪唑在高性能復合材料、生物醫藥等方面有著廣泛的用途[5]。另外,在我們研究超分子自組裝及晶體結構、多功能苯并咪唑離子液體的金屬配合物[4]和苯并咪唑類衍生物的合成[6~8]的工作中需要制備一系列苯并咪唑化合物。研究中我們發現以鄰苯二胺為原料合成苯并咪唑類化合物均為傳統合成方法[9,10],通常都在有機溶劑中進行,大量溶劑的使用會造成環境污染,不符合綠色化學清潔工藝的要求。雖然近年來有以鄰苯二胺和脂肪酸為原料的傳統無溶劑法、微波無溶劑法合成苯并咪唑類化合物的相關報道[11,12],但普遍存在消耗脂肪酸的量大、產率低、只適合微量制備等不足,不適合實驗室大量制備。
本文從實驗室制備和綠色化學角度考慮,在傳統合成方法的基礎上,首次以鄰苯二胺磷酸鹽為原料,與脂肪酸(1a~1e)經環合反應合成了五個苯并咪唑類化合物(2a~2e, Scheme 1),其結構經IR確證。
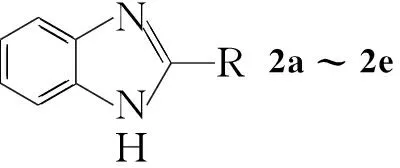

CompabcdeRHMeEtn-Prn-Bu
Scheme1
1 實驗部分
1.1 儀器與試劑
X-4型數字顯微熔點儀(溫度未校正);Nexus 670 SX型傅里葉紅外光譜儀(KBr壓片)。
所用試劑均為分析純。
1.2 2a~2e的合成(以2a為例)
在燒瓶中加入鄰苯二胺5.4 g(50 mmol)和適量磷酸,攪拌下于室溫反應1 h。減壓蒸除水分制得鄰苯二胺磷酸鹽。加入甲酸(1a) 75 mmol,回流反應7 h。傾入蒸餾水(100 mL)中,在冰水浴冷卻下用10%NaOH溶液調至pH 9~10(析出沉淀),抽濾,濾餅用無水乙醇重結晶得白色固體2a。用類似方法合成白色固體2b~2e。
2a: m.p.170 ℃~172 ℃(172 ℃~173 ℃[11]); IRν: 3 063, 2 967, 2 794, 1 619, 1 586, 1 527, 1 458, 1 276, 746 cm-1。
2b: m.p.174 ℃~175 ℃(174 ℃~176 ℃[11]); IRν: 3 059, 2 991, 2 787, 1 622, 1 555, 1 485, 1 448, 1 269, 736 cm-1。
2c: m.p.172 ℃~174 ℃(173 ℃~174 ℃[11]); IRν: 3 053, 2 974, 2 758, 1 622, 1 589, 1 543, 1 445, 1 271, 743 cm-1。
2d: m.p.155 ℃~157 ℃(156 ℃~158 ℃[11]); IRν: 3 054, 2 958, 2 769, 1 622, 1 588, 1 540, 1 455, 1 269, 746 cm-1。
2e: m.p.154 ℃~156 ℃(153 ℃~155 ℃[11]); IRν: 3 050, 2 954, 2 764, 1 623, 1 590, 1 536, 1 452, 1 273, 749 cm-1。
2 結果與討論
2.1 正交試驗優選合成2a的反應條件
以合成2a為例采用正交試驗優選反應條件。鄰苯二胺50 mmol,n(鄰苯二胺) ∶n(甲酸)(A),反應時間(B/h)和磷酸用量(C/g),通過L9(33)正交表對A, B和C三因素進行優化,正交試驗設計與結果見表1。由表1可見,各因素影響反應的次序為A>C>B,實驗的最優方案為A3B2C3。
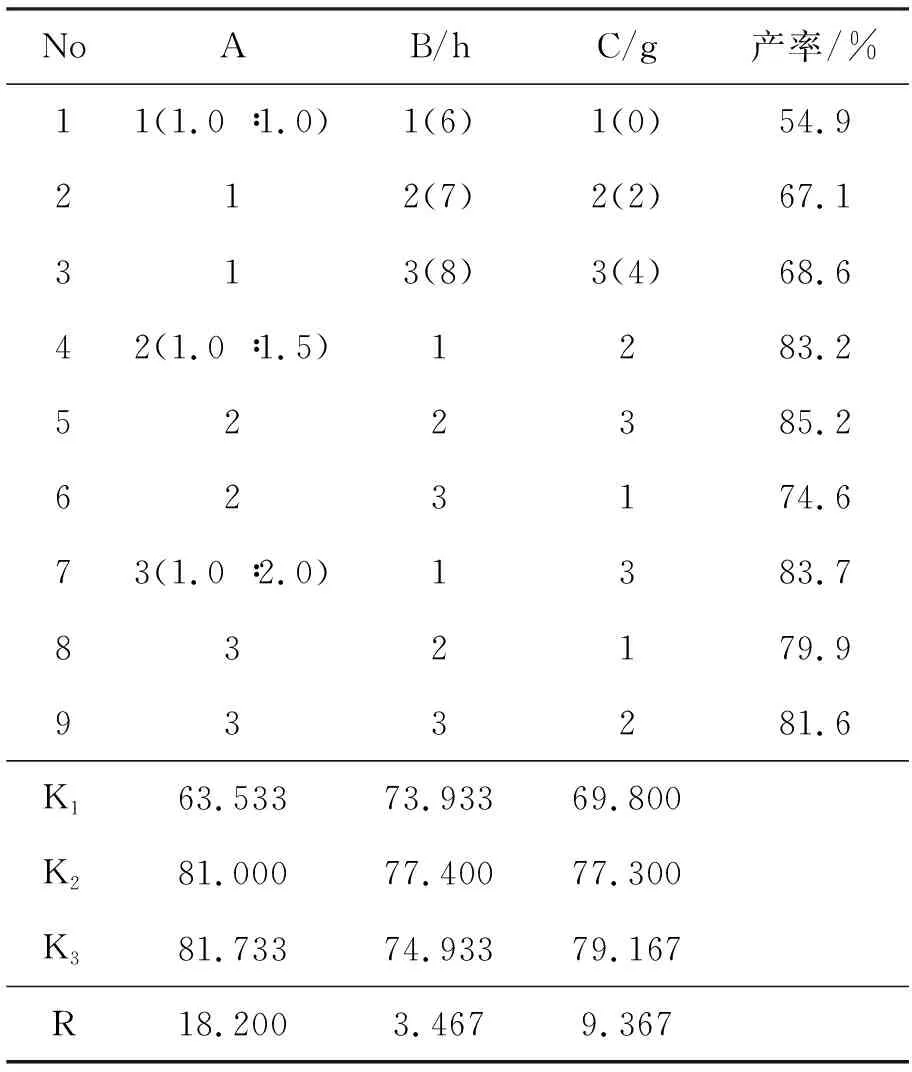
表 1 合成2a的正交試驗設計與結果*Table 1 Design and result of orthogonal experiment for synthesizing 2a
*鄰苯二胺50 mmol,n(鄰苯二胺) ∶n(甲酸)(A),反應時間(B/h),磷酸的用量(C/g),其余反應條件同1.2
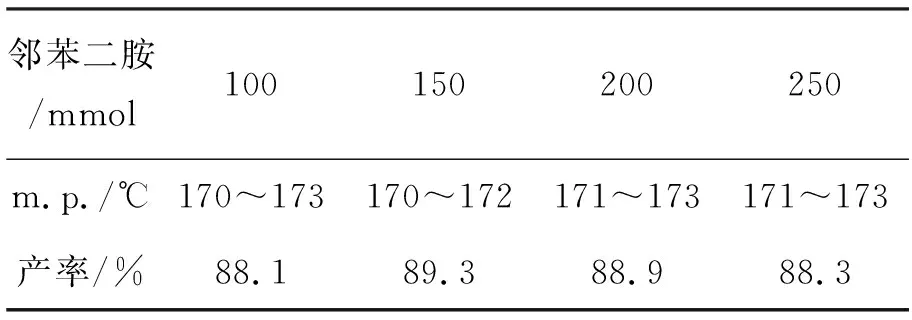
表 2 A2B2C3方案合成2a的放大實驗*Table 2 Amplification experiment for synthesizing 2a under A2B2C3
*n(鄰苯二胺) ∶n(甲酸)=1.0 ∶1.5,磷酸4 g,回流反應7 h,其余反應條件同1.2
當n(鄰苯二胺) ∶n(甲酸)分別為1 ∶1.5和1 ∶2時,其產率相差不大,從綠色化學的經濟性原則角度考慮,確定采用A2B2C3方案[n(鄰苯二胺) ∶n(甲酸)=1.0 ∶1.5,磷酸4 g,回流反應7 h],放大規模的實驗結果見表2。從表2可以看出,A2B2C3方案不僅重復性好、產率高,而且該工藝穩定可控,可以作工業化生產的進一步研究。
2.2 底物擴展
為了進一步探討A2B2C3方案的適用性及優秀性,擴展底物合成了2b~2e,結果見表3。從表3可以看出,A2B2C3方案的產率明顯比傳統無溶劑法和微波無溶劑法高,而且其熔點與文獻值接近,IR分析結果也與Scheme 1預期產物相吻合。
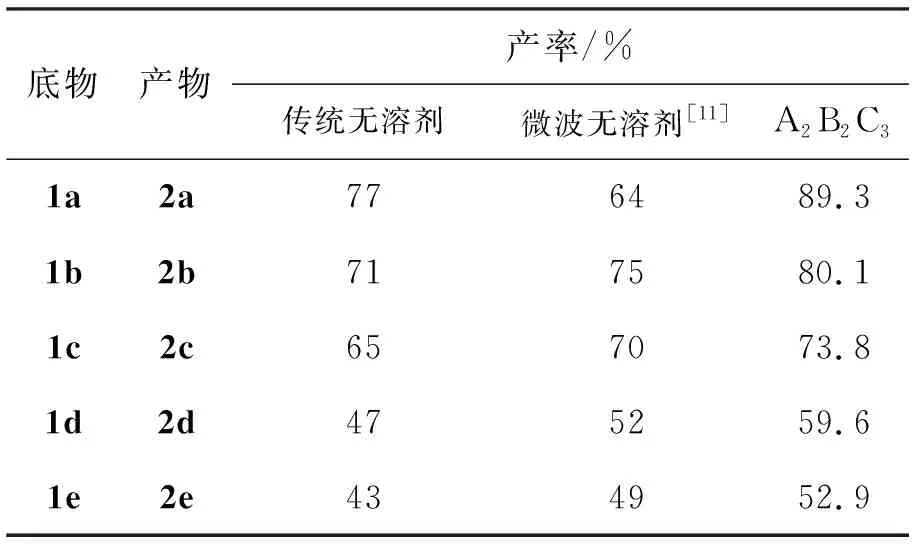
表 3 A2B2C3方案的底物擴展*Table 3 Substrate expansion under A2B2C3
*鄰苯二胺50 mmol, 其余反應條件同表2
以2d的IR分析為例:3 054 cm-1處的吸收峰是苯環的C-H峰;(1 588, 1 540, 1 455) cm-1處的吸收峰為苯環骨架伸縮振動峰,說明產物含有苯環,并且746 cm-1處的吸收峰說明了有苯環鄰位二取代;1 622 cm-1的吸收峰是N-H的彎曲振動峰,1 269 cm-1處是C-N的伸縮振動峰,所以判斷產物可能含有仲胺;(2 769, 2 958) cm-1處的吸收峰是烷基C-H的伸縮振動峰,說明產物含有烷基。IR譜圖中未發現羰基,羧基和伯胺的吸收峰,說明不含反應物。因此該合成工藝也適用于合成其他的2-烷基苯并咪唑衍生物,而且產率高。并且發現隨著碳鏈的增長其產率也隨之下降,可能的原因是碳鏈增長后,位阻增大,導致產率下降。
2.3 反應機理探討
參照文獻[12]報道,本文提出在鄰苯二胺磷酸鹽存在下可能的反應機理如Scheme 2所示。在反應過程中,鄰苯二胺磷酸鹽中的磷酸一方面保護了鄰苯二胺在反應過程中不被氧化,另一方面活化了脂肪酸中的羰基,使其很容易與氨基結合生成單酰胺。然后環合脫水,得到目標產物。
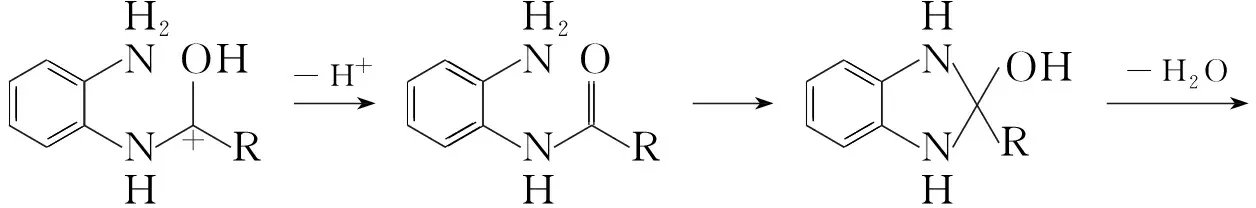
Scheme2
3 結論
對合成工藝進行了改進,采用鄰苯二胺先與磷酸在室溫下反應生成鄰苯二胺磷酸鹽,然后在無溶劑條件下與脂肪酸環合得到苯并咪唑類化合物。通過形成鄰苯二胺磷酸鹽不僅實現了無溶劑合成,而且有效的避免了鄰苯二胺被氧化,促進了環合反應的進行,提高了產率,可大量制備。通過正交設計試驗對合成工藝進行優化,旨在為苯并咪唑類化合物的實驗室研究和工業化生產奠定相應的基礎。實驗結果表明,改進后的工藝產率較文獻值[11]提高12%以上。
該工藝具有生產操作簡便、產率高、綠色環保、具有可大量工業生產潛力等優點,為苯并咪唑類化合物的在實驗室中合成提供了一種行之有效的實用方法,同時也具有較好的工業開發價值和良好的應用前景。
[1] 張璇,付玉杰,祖元剛,等. 離子液體[Etpy]·BF4催化苯并咪唑類化合物的微波合成及其抗菌活性[J].合成化學,2010,18(6):691-694.
[2] Horton D A, Bourne G T, Smythe M L. The combinatorial synthesis of bicyclic privileged structures or privileged substructures[J].Chem Rev,2003,103(3):893-930.
[3] Yang X P, Kang B S, Wong W K,etal. Syntheses,crystal structures,and luminescent properties of lanthanide complexes with tripodal ligands bearing benzimidazole and pyridine groups[J].Inorg Chem,2003,42(1):169-179.
[4] 張有明,劉勇,林奇,等. 具有新穎嵌合作用的金屬配合物的微波合成、晶體結構及性質研究[J].中國科學:化學,2011,41(5):869-877.
[5] 史子興,龐正智. 2-烷基苯并咪唑的合成及結構表征[J].北京化工大學學報,1997,24(2):27-32.
[6] 崔文輝,張有明,魏太保. 2-芳氧甲基苯并咪唑類化合物的合成[J].合成化學,2006,14(4):364-367.
[7] 魏太保,陳靖,徐蓉,等. 5-(2-芳氧甲基苯并咪唑-1-亞甲基)-1,3,4-噁二唑-2-硫酮的合成、晶體結構及生物活性研究[J].有機化學,2009,29(5):758-763.
[8] 魏太保,張治仁,師海雄,等. 微波輻射下2-芳氧甲基苯并咪唑-1-乙酰肼衍生物合成及生物活性[J].應用化學,2008,25(6):651-655.
[9] 毛鄭州,汪朝陽,侯曉娜,等. 苯并咪唑類化合物的合成研究進展[J].有機化學,2008,28(3):542-547.
[10] 唐欣,唐有根,劉小平,等. 2-取代苯并咪唑合成工藝的改進[J].廣州化學,2008,33(4):37-41.
[11] 路軍,葛紅光,白銀娟. 無溶劑微波照射下2-取代苯并咪唑的合成[J].有機化學,2002,22(10):782-784.
[12] 李英俊,劉麗軍,靳焜,等. 熔融法合成2-芳氧甲基苯并咪唑化合物[J].有機化學,2009,29(11):1825-1828.
猜你喜歡
山東冶金(2019年6期)2020-01-06 07:45:54
世界農藥(2019年2期)2019-07-13 05:55:12
科技知識動漫(2017年7期)2017-08-09 19:52:45
科技知識動漫(2017年5期)2017-05-11 21:34:16
科技知識動漫(2017年4期)2017-04-15 22:24:55
科技知識動漫(2017年2期)2017-02-06 20:59:46
科技知識動漫(2016年10期)2016-10-18 20:35:00
銅業工程(2015年4期)2015-12-29 02:48:39
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
石油化工應用(2014年8期)2014-03-11 17:40:03
