Migrastatin母核9-羥基的衍生化
2012-11-21 08:18:50鐘川龍辜玲慧吳小艾范成中
合成化學 2012年3期
鐘川龍, 辜玲慧, 吳小艾, 范成中
(1. 四川大學 a. 華西藥學院 靶向藥物教育部重點實驗室; b. 華西醫(yī)院 核醫(yī)學科,四川 成都 610041)
靶向抑制癌細胞轉移是近年來癌癥治療領域的研究熱點。Imoto等[1,2]從鏈霉菌屬培養(yǎng)液中首次分離得到Migrastatin,經生物活性測試發(fā)現(xiàn)其具有與腫瘤細胞特異性結合的能力,其構效關系研究發(fā)現(xiàn)Migrastatin十四元內酯環(huán)母核是與腫瘤細胞特異結合的有效結構單元[3]。本研究組為了借助其良好的靶向性對Migrastatin母核及其衍生物進行合成研究,希望利用未見報道的Migrastatin母核的9-羥基衍生[4~6]并通過引入示蹤元素和一系列可與示蹤元素絡合的官能團,從而獲得用于腫瘤的早期診斷和治療[4]的新型靶向試劑。
本文在Migrastatin母核全合成研究的基礎上,首先嘗試直接在Migrastatin母核9-羥基上進行衍生,但均未獲得成功。最終選擇關環(huán)之前,以2,6-二烯庚酸-5-(叔丁基二甲硅氧基)-6-甲氧基-2,4-二甲基-2,7-二烯酯(1)為起始原料,經過脫TBS保護、酯化和烯烴復分解關環(huán)反應,成功地合成了易被放射性氟(18F)代的Migrastatin母核9-甲磺酸酯[7](4, Scheme 1),總收率15.3%,其結構經1H NMR和ESI-MS表征。為實現(xiàn)其用于腫瘤早期診斷和治療研究奠定了技術基礎。
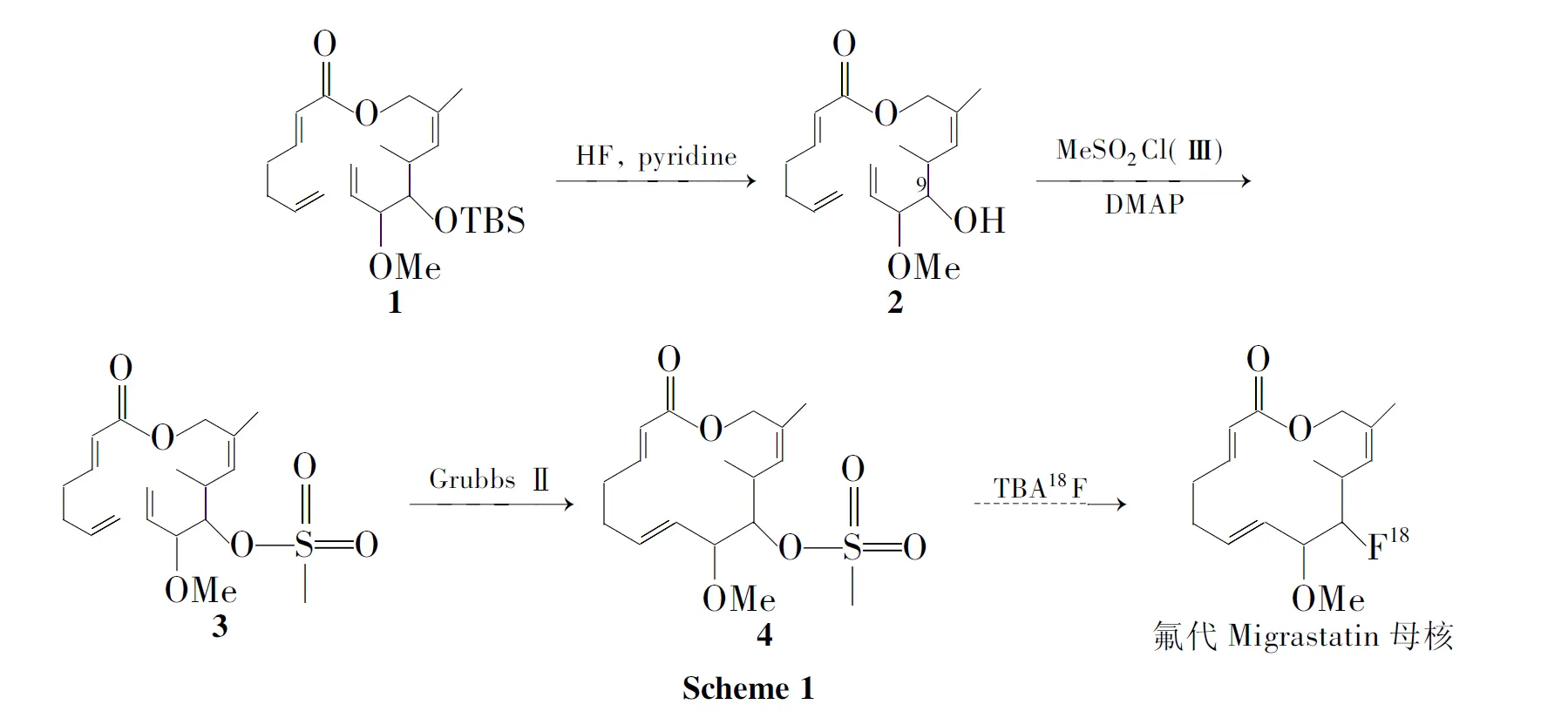
1 實驗部分
1.1 儀器與試劑
Varian Inova 400型核磁共振儀(CDCl3為溶劑,TMS為內標);Bruker Daltonics Data Analysis 3.2型質譜儀。
1按文獻[4]方法合成;THF在氮氣保護下經金屬鈉干燥重蒸;二氯甲烷在氮氣保護下經五氧化二磷干燥重蒸;甲苯在氮氣保護下由氫化鈣干燥重蒸;柱層析硅膠(硅膠H),青島海洋化工有限公司;氫氟酸吡啶鹽,Grubbs Ⅱ, MeOTMS,分析純,北京百靈威試劑有限公司;甲磺酰氯,分析純,成都愛斯特試劑有限公司。
1.2 合成
(1) 2的合成
在干燥反應瓶中加入1130 mg(0.3 mmol)的THF(9 mL)溶液,冰浴冷卻,攪拌下緩慢滴加氫氟酸吡啶鹽1.8 mL,氮氣保護下于室溫反應15 h(析出大量白色沉淀)。加入MeOTMS(甲氧基三甲基硅烷),攪拌5 min,過濾,濾餅用乙酸乙酯洗滌,合并濾液與洗滌液,減壓濃縮后經硅膠柱層析[洗脫劑:A=V(石油醚) ∶V(乙酸乙酯)=12 ∶1]純化得淡黃色油狀液體2,產率90%;1H NMRδ: 7.01~6.94(m, 1H), 5.87~5.68(m, 3H), 5.39~5.31(m, 2H), 5.08~5.00(m, 2H), 4.69~4.53(m, 3H), 3.51~3.46(m, 2H), 3.27(s, 3H), 2.73~2.65(m, 1H), 2.35~2.19(m, 4H), 1.77~1.74(m, 3H), 1.02~1.00(m, 3H)。
(2)3的合成
在干燥反應瓶中加入2100 mg(0.32 mmol)的吡啶(8 mL)溶液和DMAP(二甲氨基吡啶)3.9 mg,冰浴冷卻,攪拌下緩慢滴加稍過量的甲磺酰氯(Ⅲ,立即有白色沉淀生成),滴畢,于室溫反應2 h(TLC跟蹤)。加入少量水,分液,水層用二氯甲烷萃取,合并有機層,用無水硫酸鎂干燥,減壓濃縮后經硅膠柱層析(洗脫劑:A=12 ∶1)純化得無色油狀液體3,產率85%;1H NMRδ: 6.98~6.94(m, 1H), 5.85~5.55(m, 3H), 5.43~5.34(m, 3H), 5.07~4.99(m, 2H), 4.66~4.45(m, 3H), 3.70~3.65(m, 1H), 3.26(s, 3H), 3.08(s, 3H), 2.95~2.90(m, 1H), 2.31~2.20(m, 4H), 1.78~1.76(m, 3H), 1.04~1.01(m, 3H)。
(3)4的合成
氮氣保護,在反應瓶中加入319 mg(0.05 mmol)的甲苯(80 mL)溶液,攪拌下于120 ℃(甲苯微沸)加入Grubbs Ⅱ 8.3 mg(0.01 mmol),回流反應25 min。冷卻至室溫,過閃層硅膠柱除去甲苯,用乙酸乙酯沖洗柱子,收集液減壓濃縮后經硅膠柱層析(洗脫劑:A=10 ∶1)純化得無色油狀液體4,產率20%;1H NMRδ: 6.80~6.72(m, 1H), 5.78~5.74(d,J=16 Hz, 1H), 5.72~5.64(m, 1H), 5.48~5.33(m, 2H), 5.18~5.12(m, 1H), 4.78~4.59(m, 2H), 3.69~3.65(t,J=8.8 Hz, 1H), 3.25(s, 3H), 3.12(s, 3H), 2.51~2.21(m, 5H), 1.68(s, 3H), 0.92~0.91(m, 3H); ESI-MSm/z: 359.153 1{[M+H]+}。
2 結果與討論
2.1 Migrastatin母核9-羥基的反應活性
在全合成得到Migrastatin母核后,我們首先嘗試直接對9-羥基進行衍生。為了能引入示蹤元素和一系列可與示蹤元素絡合的官能團,分別以具有三氮中心的氨基酸(Ⅰ, Chart 1),對甲苯磺酰氯(Ⅱ)和Ⅲ為酰化劑,對Migrastatin母核9-羥基進行衍生,其反應條件及結果見表1。由表1可以看出,Migrastatin母核9-羥基的反應活性非常低,在縮合劑DCC/DMAP和EDCI/DMAP的條件下,Migrastatin與Ⅰ完全沒有反應,反應進行72 h后大量原料剩余。換用酰氯法及混酐法也同樣未檢測到酰化產物生成,如以Ⅱ或Ⅲ為酰化劑,室溫反應48 h仍然未檢測到產物。可能是由于十四元環(huán)的特殊空間構型,9-羥基處于大位阻處,其反應活性極差,無法直接完成衍生化。
本文改變合成策略,先將1的9-羥基脫TBS保護制得2; 2酯化得3;3在Grubbs Ⅱ催化下關環(huán)即完成Migrastatin母核9-羥基衍生成功地合成了4。
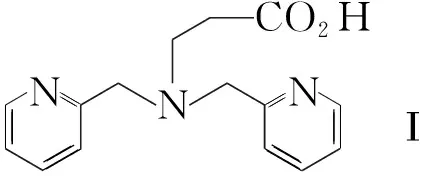
a1. A, SOCl2,回流反應2 h; 2. Migrastatin母核,CH2Cl2, 25 ℃。b1. A, 2,4,6-三氯苯甲酰氯,二異丙基乙胺,甲苯,25 ℃; 2. Migrastatin母核,吡啶,甲苯,25 ℃
2.2 3關環(huán)反應的條件優(yōu)化
以Grubbs Ⅱ試劑催化3復分解反應關環(huán)合成4,該步反應收率較低,溶劑種類、溶劑用量以及反應時間和溫度對收率都有明顯影響,其條件篩選見表2。因該反應是分子內的烯烴復分解反應,我們通過控制溶劑用量以減少分子間的副反應,從表2可以看出,當甲苯量為150 mL·(0.1 mmol)-1時,收率最高,繼續(xù)增大溶劑量并不能使收率再提高。在反應時間及溫度方面,以甲苯為溶劑,于120 ℃回流25 min即可反應完全,降低反應溫度或更換溶劑種類均不利于反應的進行。
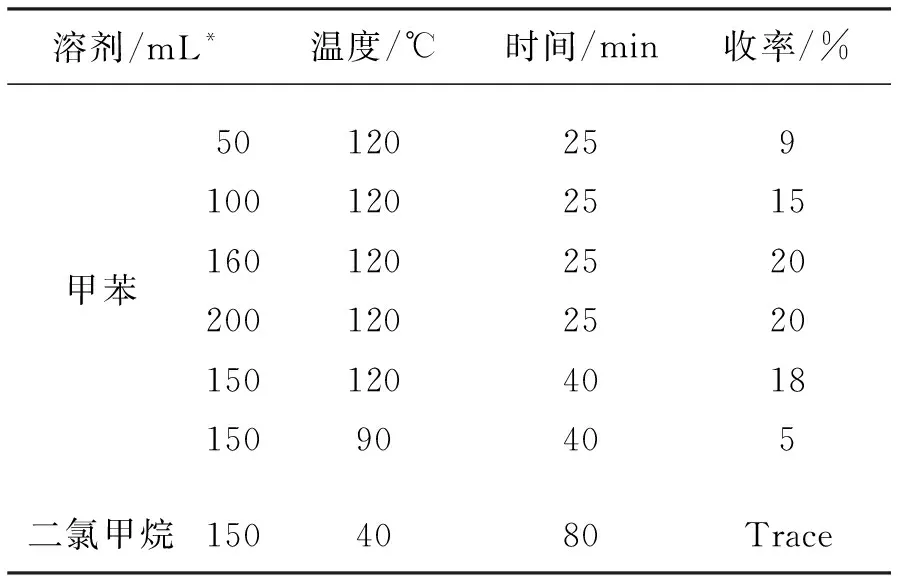
表 2 3的關環(huán)反應條件Table 2 Reactions conditions of 3 cyclization
*mL·(30.1 mmol)-1
[1] Nakae K, Yoshimoto Y, Imoto M,etal. Migrastatin a new inhibitor of tumor cell migratition from streptomycessp MK929-43F1,taxonomy,fermentation isolation and biological activities[J].J Antibiotics,2000,53(10):1130-1136.
[2] Woo E J, Starks C M, Carney J R,etal. Migrastatin and a new compoundiso-migrastatin from streptomyces platensis[J].J Antibiotics,2002,55(2):141-146.
[3] Njardarson J T, Gual C, Shan D,etal. Discovery of potent cell migration inhibitors through total synthesis:Lessons from structure-activity studies of (+)-Migrastatin[J].J Am Chem Soc,2004,126(4):1038-1040.
[4] 唐柏楊,吳小艾,謝蔣平,等. Migrastatin母核的全合成[J].華西藥學雜志,2011,26(2):103-107.
[5] Ebastien R, Janine C. Synthesis of Migrastatin and its macrolide core[J].Tetrahedron,2007,63(26):918-5929.
[6] Njardarson J T, Gual C, Shan D,etal. The Migrastatin family:Discovery of potent cell migration inhibitors by chemical synthesis[J].J Am Chem Soc,2004,126(36):11326-11337.
[7] Zhu L, Liu Y J, Pl?ssl K,etal. An improved radiosynthesis of [18F]AV-133:A PET imaging agent for vesicular monoamine transporter 2[J].Nuclear Medicine and Biology,2010,37:133-141.
