通脈抗栓顆粒質量標準研究
2012-11-09 09:39:30李成網
中國醫藥指南 2012年9期
張 潔 白 娟 李成網*
(安徽省醫學科學研究院,安徽 合肥 230061)
通脈抗栓顆粒是根據臨床經驗方開發的六類中藥新藥,已獲國家藥品食品監督管理局頒發的臨床批件(批件號:2007L05140),由當歸、川芎、葛根、人參葉等中藥組成,具有益氣養陰,活血通脈等功效。用于腦血栓癥見頭暈目眩,頭痛,肢體麻木或強急,氣短乏力,便干便秘,口干口渴,舌質紅或質暗淡等氣陰兩虛, 瘀阻腦絡證。采用薄層色譜法對其中的當歸、川芎、麥冬、人參葉進行定性鑒別,采用HPLC法對葛根中的有效成分葛根素進行含量測定,可有效控制通脈抗栓顆粒的質量。
1 儀器、試藥與樣品
Waters 600型高效液相色譜儀(美國Waters公司);UV 996型紫外檢測器;AD-2型百萬分之一電子天平(美國PE公司);BP210S型萬分之一電子天平(德國賽多利斯);AS3120型超聲清洗器(天津奧特賽恩斯);葛根素為對照品(批號0752-9806供含量測定用);人參皂苷Rb1對照品(批號704—8903);人參皂苷Re對照品(批號754-9002);人參皂苷Rg1對照品(批號0703-200119);當歸對照藥材(批號120927-200310);川芎對照藥材(批號120918-200306);麥冬對照藥材(批號121013-200607)均購于中國藥品生物制品檢定所。甲醇、磷酸為色譜純,水為重蒸水,其他試劑均為化學純。通脈抗栓顆粒由安徽省華康醫藥科技開發有限責任公司提供(規格:每袋裝8g)。
2 方法與結果
2.1 薄層色譜鑒別
2.1.1 當歸川芎的鑒別[1]
取本品15g,研細,加乙醚150ml浸漬1h后,超聲處理5min,濾過,濾液揮干,殘渣加醋酸乙酯0.5mL使溶解,作為供試品溶液。另取當歸對照藥材和川芎對照藥材各1g,分別加乙醚50mL,浸泡1h,超聲處理5min,濾過,濾液揮干,殘渣加醋酸乙酯2mL使溶解,分別作為對照藥材溶液。照薄層色譜法試驗,分別吸取當歸和川芎對照藥材溶液3μL,供試品溶液10μL,分別點于同一硅膠G薄層板上,以正已烷-醋酸乙酯(9∶1)為展開劑,展開,取出,晾干,置紫外光燈(365nm)下檢視。供試品色譜中,在與對照藥材色譜相應的位置上,顯兩個相同顏色的熒光斑點。陰性對照無干擾。見圖1。

圖1 當歸川芎鑒別薄層色譜圖
2.1.2 麥冬的鑒別[2]
取鑒別(1)項下乙醚提取后的藥渣,加乙醇100mL回流1h,濾過,濾液回收乙醇至干,加3%硫酸溶液10mL置沸水浴加熱回流2h,用3%氫氧化鈉溶液調pH至中性,水浴蒸干,殘渣加氯仿2mL使溶解,作為供試品溶液,另取麥冬對照藥材2g,加乙醇20mL回流1h,同法制成對照藥材溶液。照薄層色譜法試驗,吸取對照藥材溶液10μL和供試品溶液10μL,分別點于同一硅膠G薄層板上,以正己烷-醋酸乙酯(1∶1)為展開劑,展開,取出,晾干,噴以10%硫酸溶液,于105°C加熱至斑點顯色清晰。供試品色譜中,在與對照藥材色譜相應的位置上,顯相同顏色的斑點。陰性對照無干擾。見圖2。
2.1.3 人參葉的鑒別[3,4]

圖2 麥冬鑒別薄層色譜圖
取本品5g,研細,加甲醇50mL,超聲處理20min,濾過,濾液蒸干,殘渣加水25mL使溶解,濾過,濾液加水飽和正丁醇提取3次,每次25mL,合并正丁醇提取液,用正丁醇飽和的氨試液洗3次,每次25mL,棄去氨試液,正丁醇液水浴蒸干,殘渣加甲醇2mL使溶解,作為供試品溶液。另取人參皂苷Rb1、Re、Rg1對照品,分別加甲醇制成每1mL各含0.5mg的溶液,作為對照品溶液。照薄層色譜法試驗,吸取供試品溶液5~10μL,對照品溶液3~5μL,分別點于同一硅膠G薄層板上,以正丁醇-醋酸乙酯-水(4∶1∶5)的上層液為展開劑,展開,取出,晾干,噴以10%硫酸乙醇溶液,在105℃加熱至色斑清晰,供試品色譜中,在與對照品色譜相應的位置上,顯相同顏色的斑點。陰性對照無干擾。見圖3。

圖3 人參葉鑒別薄層色譜圖
2.2 葛根素的含量測定
葛根為方中君藥,其主要成分為葛根素。用高效液相色譜法測定中藥制劑中葛根素的含量的報道很多[5,6]。在參考文獻的基礎上,為了有效地控制本品的質量,建立了本品的葛根素的高效液相色譜法的測定法,經方法學考察,本法簡便、準確、可靠、重現性好,可有效控制本品質量,其方法學考察如下:
2.2.1 色譜條件與系統適用性試驗
2.2.1.1 對照品溶液的制備
精密稱取葛根素對照品適量,加30%乙醇制成每1mL含60μg的溶液,即得。
2.2.1.2 供試品溶液的制備
取本品,研細(過4號篩),取0.5g,精密稱定,置具塞錐形瓶中,精密加入30%乙醇50mL,密塞,稱定重量,超聲處理30min,放冷,再稱定重量,用30%乙醇補足減失的重量,搖勻,濾過,取續濾液,即得。
2.2.1.3 陰性對照溶液的制備
取缺葛根的陰性制劑,按供試品溶液制備項下的方法依法制備陰性對照溶液。
2.2.1.4 色譜條件的選擇
色譜柱:Shim-Pack CLC ODS柱(150×4.6mm,5μm);流動相:甲醇-0.1%磷酸溶液(24∶76);檢測波長:250nm;柱溫:30℃;流速:1.0mL/min。在此條件下,葛根素與其它成分均能很好地分離,供試品中葛根素保留時間與對照品一致,且陰性對照無干擾,結果見圖4。
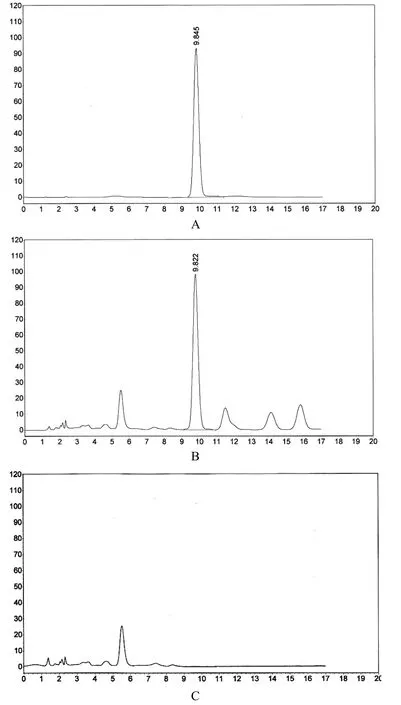
圖4 對照品、樣品、陰性樣品HPLC色譜圖
2.2.5 線性關系考察
精密吸取葛根素對照品溶液(0.21744mg/mL)1.0、2.0、3.0、4.0、5.0mL分別置10mL量瓶中,加30%乙醇稀釋至刻度,搖勻。分別精密吸取20μL,依次注入液相色譜儀中,按上述色譜條件測定,峰面積分別為:1516068、2931786、4409670、5889355、7336747。以葛根素的濃度為橫坐標,相應的峰面積積分值為縱坐標,計算,得線性回歸方程為:A=67140C+37047 r=0.99997(n=5)。表明葛根素在21.744~108.72μg/mL范圍內呈良好的線性關系。
2.2.6 供試品精密度試驗
取按供試品溶液制備法制備的供試液(批號040805),按上述色譜條件注入液相色譜儀,連續進樣5次,記錄葛根素峰面積分別為:5179674、5275425、 5276877、5392464、5317029,RSD=1.46%,表明精密度良好。
2.2.7 穩定性試驗
將按供試品溶液制備法制備的供試液(批號040805)于0、1、2、4、6、8h按上述色譜條件重復測定一次,記錄葛根素峰面積分別為:5463520、5417945、5489535、5464131、5471071、5561982,RSD=0.87%。表明供試液在8h內穩定。
2.2.8 重復性試驗
取同一批號樣品(批號040805)共5份,按供試品溶液制備法制備供試液,精密量取該溶液和對照品溶液各20mL,按上述色譜條件注入液相色譜儀,記錄色譜圖,計算葛根素含量分別為65.5728mg/袋,65.7864mg/袋,64.1368mg/袋,65.0384mg/袋,64.9488mg/袋,RSD=0.98%。表明方法重復性良好。
2.2.9 回收率試驗
精密量取已知含量(65.0968mg/袋)的樣品適量,分別精密加入葛根素對照品適量,按正文含量測定項下的方法依法制備供試品溶液并測定,結果見表1。

表1 回收率試驗結果(n=6)
2.2.10 樣品測定
對3批中試樣品按供試品溶液制備方法制備樣品溶液,分別精密吸取樣品溶液和對照品溶液各20mL,注入液相色譜儀,按外標法以峰面積計算含量,結果見表2。

表2 樣品含量測定結果
3 討 論
3.1 本方為中藥復方制劑,采用薄層色譜鑒別方法對方中的每一味中藥均進行了鑒別,并進行了陰性對照試驗,結果其方法簡便且專屬性強,可有效控制本品的質量。
3.2 關于葛根中葛根素的含量測定方法,文獻報道很多,但在本制劑中未見報道,對流動相進行了選擇,先后比較了甲醇-水(25∶75)、甲醇-水(22∶78)、甲醇-0.1%磷酸溶液(22∶78)、甲醇-0.1%磷酸溶液(24∶76)。結果以甲醇-0.1%磷酸溶液(24∶76)為流動相最佳。
3.3 對供試品溶液制備的超聲提取時間也做了考察,分別比較了超聲處理10、20、30、40min的效果。結果表明,超聲提取30min已能提取完全,因此超聲提取時間選擇30min為佳。
[1]中華人民共和國衛生部藥典委員會編.中華人民共和國藥典[S].2000年版一部.北京:化學工業出版社,2000:433.
[2]余伯陽.不同麥冬假葉樹皂甙元的含量比較[J].中國藥科大學學報,1987,18(2):117.
[3]中華人民共和國衛生部藥典委員會編.中華人民共和國藥典[S].2000年版一部.北京:化學工業出版社,2000:6.
[4]苗明三,李振國.現代實用中藥質量控制技術[M].北京:人民衛生出版社,2000:36.
[5]楊小平.RP-HPLC法測定感冒清熱顆粒中葛根素的含量[J].藥物分析雜志 2001,21(5):331.
[6]劉建峰.HPLC法測定舒心片中葛根素含量[J].西北藥學雜志,2000,15(5):1990.
