LiFePO4/C|石墨鋰離子電池的高溫循環性能
2012-07-05 01:39:44賴旭倫趙豐剛
電池 2012年4期
關鍵詞:體系
許 瑞,賴旭倫,趙豐剛,吳 凱
(東莞新能源科技有限公司,廣東東莞 523808)
采用水基膠粘劑并用水代替N-甲基吡咯烷酮(NMP)作為溶劑,開發水基體系磷酸鐵鋰(LiFePO4)正極,具有價格方面的優勢。對相同的材料而言,水基體系與以NMP為溶劑的油基體系相比,在60℃下的1 C循環性能略差,而45℃及常溫(23±2℃)下的性能基本上沒有差別。高溫循環性能差,也是限制 LiFePO4材料應用的主要原因之一[1]。
LiFePO4/C|石墨體系動力電池的高溫循環性能不理想[2-3]。K.Amine等[2]發現,陽極極化的增加較快,且在高溫下,正極中的Fe3+溶解并沉積在陽極表面,還會進一步還原,生成金屬Fe顆粒;若將有可能分解生成HF的 LiPF6鹽換成LiBOB,電池中負極Fe的沉積量會急劇下降。水基LiFePO4/C|石墨體系鋰離子電池高溫循環性能衰減較快的原因有:①Fe3+溶出有可能會加劇高溫循環衰減;②采用水基體系正極,在電芯制備過程中,水分可能脫除不干凈,會加劇 LiPF6分解生成HF,進而加速鐵Fe3+的溶解和沉積。
N.Dupré等[3]發現,在 LiFePO4表面包覆碳層,可對小顆粒、大比表面積的納米LiFePO4與電解液接觸的表面進行保護,阻止Fe3+的溶出,在高溫貯存或循環時,正極表面會形成電子導通性較差的有機層。
不同廠家使用的碳源及包覆工藝不同,效果差別大,因此需要對水基LiFePO4/C|石墨體系鋰離子電池高溫貯存及循環時的實際行為進行量化分析。本文作者對LiFePO4/C|石墨體系鋰離子電池進行分析,取得高溫循環性能量化的結果,以對循環衰減的機理作進一步的分析、研究和驗證。
1 實驗
1.1 粉體分析
用JEM-2010 HR型透射電子顯微鏡(TEM,日本產)對LiFePO4/C(東莞產,碳含量3.2%,工業級)進行分析,觀察LiFePO4及表面碳層的包覆情況。
1.2 電池的制備
以聚偏氟乙烯(PVDF,美國產,電池級)、導電碳 SP(瑞士產,電池級)、LiFePO4/C、聚乙烯醇(PVA,英國產,工業級)為原料,按溶劑型配方 m(PVDF)∶m(SP)∶m(LiFePO4)=5∶5∶90 和水基配方 m(PVA)∶m(SP)∶m(LiFePO4)=5∶5∶90分別制備 LiFePO4漿料,并按 0.02 g/cm2的量涂覆于處理過[4]的16μ m厚的鋁箔(東莞產,電子級)上,制備正極片,極片密度為2.1 g/cm3。將羧甲基纖維素鈉(CMC,東莞產,食品級)、導電碳SP、丁苯橡膠(SBR,東莞產,工業級)與人造石墨(東莞產,電池級)按質量比2∶2∶2∶94混合制漿,并涂覆0.01 g/cm2于10 μ m厚的銅箔(東莞產,電子級)上,制備負極片,極片密度為 1.5 g/cm3。 以 Celgard M824膜(16 μ m厚,美國產)為隔膜,1 mol/L LiPF6/EC+PC+DEC(體積比 1∶1∶1,>99.99%,東莞產)為電解液,鋁塑膜為密封包裝件,按設計的3.0 Ah容量,采用本公司的卷繞工藝,制備尺寸為109 mm×101 mm×30 mm的方形軟包裝電池。
將漆包銅絲整理成平行的單根、Φ=32 μ m 的銅絲,將銅絲一端長約30 mm,浸入 1 mol/L H2SO4(深圳產,AR)中浸泡10~20 min,取出后用去離子水清洗,再用無水乙醇(廣東產,AR)浸泡,洗凈殘留的物質。為觀察循環過程中正、負極的變化,在制備電池前的工序中,取出未注液頂封的電池,在隔膜中插入處理后的銅絲(東莞產,電子級),制成三電極電池,其他制備工序與正常電池相同。鋁塑膜熱封后,外露的銅絲用鎳片(0.1 mm×4.0 mm,東莞產,電子級)轉接焊,并用Kapton高溫膠帶(美國產,電子級)固定。
將電池按生產流程進行活化和容量測試,取合格且性能一致的電池,在LIP-3AHB01型鋰離子電池高溫化成系統(杭州產)上進行60℃、1 C循環,電壓為2.00~3.65 V。
為研究水基正極片的電化學阻抗譜(EIS),用模具沖切極片、隔膜,預留對稱的極耳區(長25.0 mm,寬 4.0 mm)并焊接0.1 mm×4.0 mm的鋁片,灌注電解液,制備對稱電池。
為考察漿料貯存后極片的擴散性能是否有變化,以及驗證文獻[3]中的結果,將制備極片的漿料靜置30 d,再涂覆、干燥,并按相同的步驟制備電極與對稱電池,進行分析。
1.3 電化學性能分析
將電池按照設計的1 C循環程序循環,每循環100~200次后,再以0.2 C小電流放電150 min[放電深度(DOD)為50%],放完電后,將待測電池拆解,并將開路電壓為3.2~3.3 V電池的正極與銅絲電極接入2000型電池測試系統(武漢產)的相應電極接頭,用小電流(20~50 μ A)對銅絲鍍鋰 2 h,將鍍完鋰的銅電極作為鋰參比電極。
用Im 6eX電化學工作站(德國產)對上述鍍鋰的三電極電池及對稱電池進行EIS測試,頻率為30 mHz~500 kHz。
EIS測試完畢后,將三電極電池置于恒溫箱中,以正極及參比電極作為監控對象,在BT 2000型電池綜合測試儀上(美國產)進行一次60℃下的0.2 C循環。
取新鮮的負極片及循環測試過程中的LiFePO4/C|石墨體系電池,放電至2.0 V,拆解并取出負極片,刮取2~5 g未洗滌的電極粉末,在105℃下烘干2 h后,進行消解處理[5],再用Thermal iCAP 6300型電感耦合等離子發射光譜(ICPOES)儀(美國產)進行 ICP-OES測試[5],分析Fe3+的含量。
2 結果與討論
2.1 粉體的TEM分析
圖1為LiFePO4/C的TEM圖。
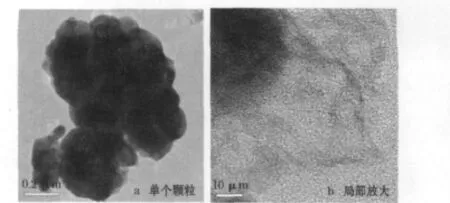
圖1 LiFePO4/C的TEM圖Fig.1 Transmission electron microscopy(TEM)photographs of LiFePO4/C
從圖1可知,LiFePO4/C是100 nm大小的一次顆粒表面包覆導電碳的結構,并由幾十個一次LiFePO4顆粒團聚成為二次顆粒,二次顆粒的粒徑約為 1 μ m,一次 LiFePO4顆粒的表面包覆了不均勻的導電碳層。包覆的碳層為無定形碳,碳層為原位生成,厚度在20 nm以上。
2.2 對稱電池的EIS
LiFePO4/C極片對稱電池的EIS見圖2。
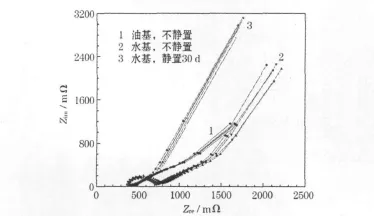
圖2 LiFePO4/C極片對稱電池的電化學阻抗譜(EIS)Fig.2 Electrochemical impedance spectra(EIS)of symmetrical cell using LiFePO4/C electrode
從圖2可知,與油基 LiFePO4/C)相比,水基LiFePO4/C極片的電阻(實部)相近,但有一個明顯的半圓,且第 2個半圓簡化成一條直線,說明水基LiFePO4/C極片的鋰鹽電解質(LiPF6)擴散系數較大,電解質在其中的擴散較差。水基漿料攪拌后靜置30 d再制備的對稱電池,半圓環變弱,第2個半圓近似消失,與油基極片接近,此現象需要進一步研究。
2.3 三電極電芯監測及分析
2.3.1 EIS分析
對循環不同階段的三電極電池的正極與參比電極、正極與負極以及負極與參比電極分別進行50%DOD放電,再進行EIS分析,結果見圖 3。
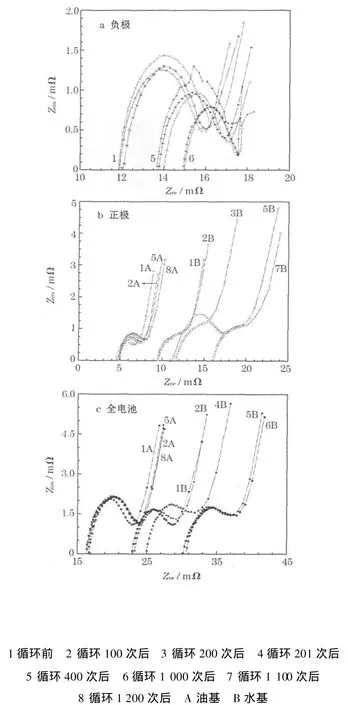
圖3 不同循環階段電池的EISFig.3 EIS of cell at different cycle state
從圖3a可知,兩種LiFePO4/C|石墨體系鋰離子電池的負極EIS在循環過程中有變化,變化趨勢幾乎相同,且隨著循環的進行,譜圖中半圓環幾乎不變,而直線區域逐漸變短,接近消失,表明LiFePO4/C|石墨體系鋰離子電池的負極界面有變好的趨勢;從圖3b可知,油基體系的正極阻抗譜變化較大,而水基體系的正極EIS呈增大趨勢(實部從9.5 mΩ增加到16.2 mΩ),表明正極極化增大是水基體系全電池阻抗增大的主要原因,與文獻[2]的報道略有不同。水基正極與油基正極主要差別在于膠粘劑,通常,油基正極用的膠粘劑為PVDF等,而本實驗選用的膠粘劑為PVA。
從圖3c可知,正、負極的EIS與全電池的EIS有加和性,且引起上述變化的最主要因素是正極,負極無論所對應的正極是水基體系還是油基體系,均變化不大。從圖3可知,水基體系正極的內阻較油基體系約大100%。
2.3.2 正/負極放電曲線對比
為了進一步研究循環過程中正、負極的變化,以金屬鋰為參比電極,對循環過程中不同階段的三電極電池的正、負極的電位進行分析,結果見圖4。
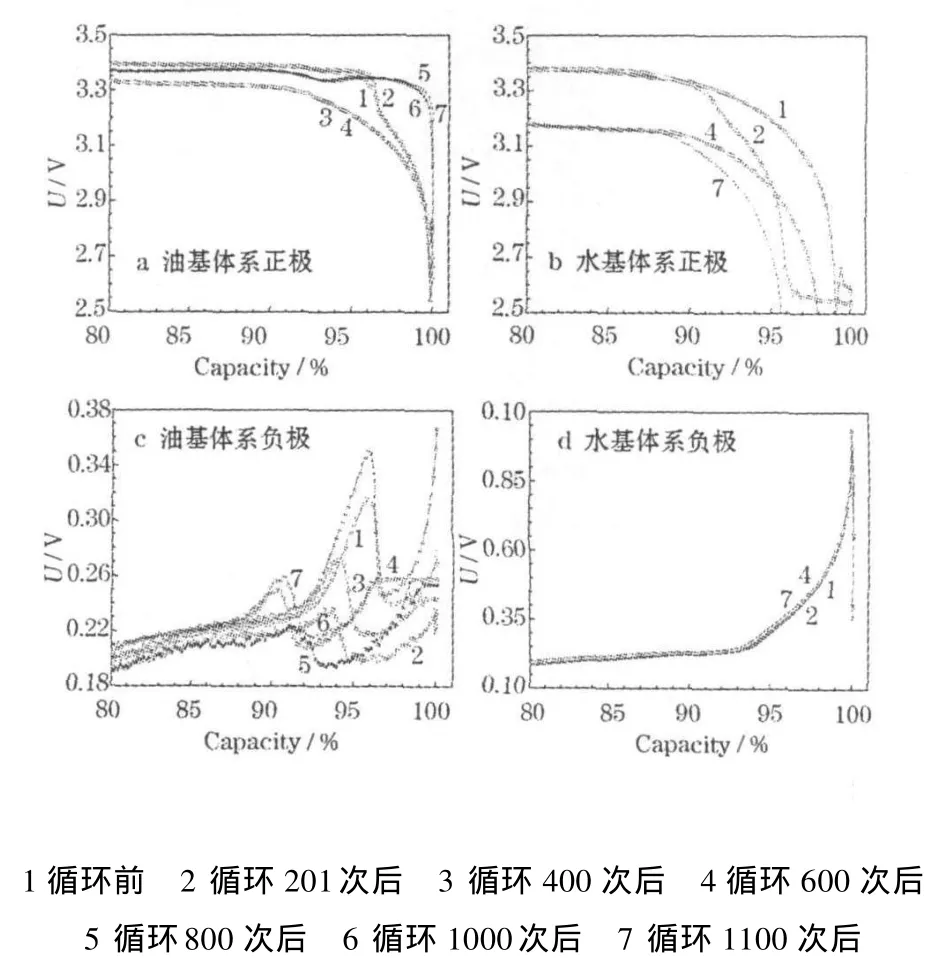
圖4 不同循環階段三電極電池正、負極的電位Fig.4 Potential of anode and cathode of tri-electrode cells at different cycle state
從圖 4可知,不同循環階段,負極的電位變化不大,而正極有較明顯的變化。水基體系的正極電位逐漸下降,若負極電位不變化,表明正極的極化是導致LiFePO4/C|石墨體系鋰離子電池高溫循環性能下降的主要原因。在截止電位一定的情況下,電池的放電容量會減少,即循環容量衰減過快。
在循環過程中,油基 LiFePO4/C|石墨體系鋰離子電池的負極界面相對比較穩定,但容量平穩下降,正極極化有增大的趨勢,水基體系正極的變化較為明顯。
2.3.3 循環過程中負極片Fe3+含量的變化
LiFePO4/C|石墨體系鋰離子電池循環的容量保持率及循環過程中負極Fe3+含量的變化見圖5。
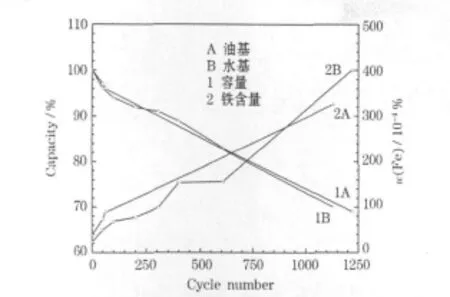
圖5 LiFePO4/C|石墨體系鋰離子電池負極的鐵含量Fig.5 Fe3+contents of LiFePO4/C|graphite system Li-ion batteries
從圖5可知,LiFePO4/C|石墨體系鋰離子油基三電極電池在60℃時1 C循環1 100次,容量保持率約為70%,而水基LiFePO4/C|石墨體系鋰離子三電極電池容量保持率為70%所對應的循環次數約為1 000次。兩種LiFePO4/C|石墨體系鋰離子三電極電池的Fe3+溶出隨著循環次數的增加均呈線性上升的趨勢,且速率接近,表明膠粘劑對Fe3+的溶出反應沒有影響;在電池循環過程中,Fe3+的溶出一直進行,與文獻[3]報道的有所不同。
3 結論
研究了水基(油基)LiFePO4/C|石墨體系鋰離子電池在60℃時的1 C循環性能。在相同的容量保持率(70%)下,水基體系較油基體系的循環次數約少100次;Fe3+的溶出速度與膠粘劑無關。
水基LiFePO4/C|石墨體系鋰離子電池正極極化逐漸增大;在相同配比的條件下,水基膠粘劑制備的正極片較油基膠粘劑制備的正極片擴散系數大,是高溫循環性能較差的主要原因。
[1]CHE Hai-ying(車海英),YANG Jun(楊軍),WU Kai(吳凱),et al.二(三氟甲基磺酰)亞胺鋰對磷酸鐵鋰正極高溫行為的影響[J].Acta Chimica Sinica(化學學報),2011 69(11)1 287-1 292.
[2]Amine K,Liu J,Belharouak I.High-temperature storage and cycling of C-LiFePO4/graphite Li-ion cells[J].Electrochem Commun,2005,7(7):669-673.
[3]Dupr é N,Jean-Frédé ric M,Jeremy D,et al.Aging of the LiFePO4positive electrode interface in electrolyte[J].J Power Sources,2010,195(21):7 415-7 425.
[4]MEI Ming(梅銘),LAI Xu-lun(賴旭倫),HE Li-ping(何麗萍),et al.鋰離子電池用水基印刷導電油墨的制備[J].Battery Bimonthly(電池),2012,42(3):132-135.
[5]NI Wei-hong(倪偉紅),CHEN M ei-chun(陳美春),JIA Yan-bo(賈彥博).微波消解ICP-OES法同時測定化妝品中 Pb,As,Hg.[J].Chinese Journal of Analysis Laboratory(分析實驗室),2008,27(12):326-328.
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
