3- 取代劍麻皂素的合成
2012-04-01 01:57:12甘春芳林啟福陸秀蓮崔建國
化工技術與開發 2012年10期
甘春芳,劉 亮,林啟福,陸秀蓮,崔建國
(廣西師范學院化學系,廣西 南寧 530001)
從自然界存在的生物體中分離出具有生理活性的天然產物[1],對它們進行結構分析、藥理研究和臨床試驗[2~3],從而發現能夠治療某種疾病的藥物,是人們發現新藥的一條主要途徑。
劍麻皂素(Tigogenin)是從生產劍麻纖維傳統產品之后廢棄的麻汁和麻渣中提取的天然植物皂苷元,是合成甾體激素類藥物的醫藥中間體和重要原料,廣泛應用于腎上腺皮質激素、性激素及蛋白同化激素3大類200多種藥物的制造[4~5]。
從文獻上看,目前對劍麻皂素的研究,大多數都是對其醚鍵進行改造得到多種具有良好藥用價值的化合物。本課題組在研究中發現,在甾核的3- 位引入肟基、腙基等不同的官能團時,這些甾體類化合物具有良好的細胞活性和生理藥性[6~7]。劍麻皂素是天然存在的具有生理活性的物質,其具有甾核的基本結構,那么在其3- 位引入不同含氮的官能團是否也具有較好的抗腫瘤活性呢?因此,本文試圖通過改造劍麻皂素的3- 位羥基成為不同的含氮基團,希望得到一些有用的具有特殊生理活性的化合物,為新藥的研發提供有用的理論參考。
1 實驗部分
1.1 儀器與試劑
Nicolet Avater FT-IR 360傅立葉紅外光譜儀,Bruker 300 MHz超導核磁共振儀,BS200S電子天平,以及實驗室常用的一些儀器。實驗試劑均為分析純。
1.2 合成路線
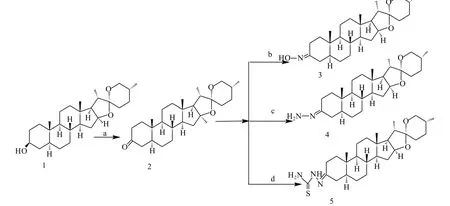
試劑與條件:(a)Collins/CH2Cl2;(b)NaAc·3H2O,NH2OH·HCl;(c)NH2-NH2·H2O/CH3CH2OH;(d)NH2NHCSNH2/CH3CH2OH。
1.3 實驗步驟
1.3.1 三氧化鉻吡啶(Collins reageat)的制備
在冰浴條件下,將10g CrO3(0.1mol)緩慢加入過量的吡啶(約20mL)中,不斷攪拌,直至生成的黃色晶體轉變成深紅色。抽濾,石油醚洗滌,干燥,放入真空干燥箱中抽干備用。
1.3.2 3- 羰基 - 劍麻皂素(2)的制備
在100mL反應瓶中加入202mg(0.486mmol)劍麻皂素,加入30mL CH2Cl2溶解后,攪拌條件下,一次性加入526mg(2.04mmol)Collins試劑,溶液迅速變成深褐色,反應在室溫條件下攪拌進行。TLC監測(展開劑:V乙酸乙酯∶V石油醚=1∶4),反應大約 30h后沒有原料點,停止反應。反應物傾進硅膠短柱,乙酸乙酯洗脫,減壓蒸出溶劑,使用0.038~0.048mm硅膠柱層析分離(洗脫劑:V乙酸乙酯∶V石油醚=1∶4),得到白色固體142mg。產率:72%,θmp:214~215℃。該化合物的光譜數據: IR(KBr,υ/cm-1): 2945,1732,1437,1380,1249,1053;1H NMR(CDCl3,300MHz)δ:4.37~4.39(1H,m,C16-H), 3.321~3.391(2H,m,C26-H),1.106(3H,s, 27-CH3), 0.954(3H, d,J=6.6 Hz, 21-CH3), 0.780(s,6H,18-CH3and 19-CH3);13C NMR(CDCl3,75MHz) δ:213.0(3-C), 109.2(22-C),80.7(16-C),66.8(26-C),62.1(17-C),56.1(14-C),53.7(9-C),46.6(5-C), 44.7(4-C),41.6(20-C),40.5(13-C),39.9(12-C),38.5(1-C),38.1(2-C),35.7(10-C), 35.0(8-C),31.8(15-C),31.7(23-C),31.3(7-C),30.3(24-C),28.8(6-C),28.7(25-C),21.2(11-C),17.1(27-C),16.5(18-C),14.5(19-C),11.5(21-C)。
1.3.3 3- 肟基 - 劍麻皂素(3)的制備
86mg(0.208mmol)化合物2溶于95%乙醇中,溶解完全后加入28mg(0.208 mmol)NaAc·3H2O,攪拌10min 后,加入 19mg NH2OH·HCl(0.273mmol),保持水浴溫度為60℃,TLC監測(V乙酸乙酯∶V石油醚= 1∶4),反應28h后沒有原料點,停止反應。減壓蒸去大部分乙醇,得到白色固體,加入適量的水,用乙酸乙酯(10mL×3)萃取,有機相用飽和食鹽水洗至中性,無水硫酸鈉干燥,過濾,減壓蒸去溶劑,粗產品使用硅膠柱層析分離(洗脫劑:V乙酸乙酯∶ V石油醚=1∶4), 得到白色固體80mg。產率90%,θmp∶249~250℃。化合物 3 的光譜數據:IR(KBr,υ/cm-1):3383,2921,1642,1552,1446,1385,1344,1242;1H NMR(CDCl3,300MHz) δ:8.697(1H,s,N-OH),4.39~4.41 (1H,m,C16-H),3.37~3.50(2H,m,C26-H),0.970(3H,br s,21-CH3),0.916(3H,s,27-CH3),0.784(6H,br s,18-CH3and 19-CH3);13C NMR(CDCl3,75MHz) δ:160.5(3-C),109.2(22-C),80.8(16-C),66.8(26-C),62.1(17-C),56.2(14-C),54.0(9-C), 46.4(5-C),45.3(4-C),41.6(20-C),40.5(13-C),40.0(12-C),38.3(2-C),37.1(1-C), 36.3(10-C),35.0(8-C),32.0(15-C),31.7(23-C),31.4(7-C),30.3(24-C),28.8(6-C), 28.5(25-C),20.4(11-C),17.2(18-C),16.5(27-C),14.5(19-C),11.4(21-C)。
1.3.4 3- 腙基劍麻皂素(4)的制備
170mg(0.411mmol)3- 羰基劍麻皂素溶于30mL無水乙醇中,溶解完全后滴入3~4滴冰醋酸調節溶液的pH值為3~5,然后加入45μL水合肼(80%)溶液,保持水浴溫度為60℃,TLC監測(V乙酸乙酯∶V石油醚=1∶4)反應,大約1h后沒有原料點,停止反應。減壓蒸去無水乙醇,得到白色固體,加入適量CH2Cl2溶解,再加入適量水,然后用二氯甲烷(10mL×3)萃取。有機相用飽和食鹽水洗至中性,無水硫酸鈉干燥,過濾,減壓蒸去溶劑,粗產品使用0.048~0.075mm硅膠柱層析分離(洗脫劑:V乙酸乙酯∶V石油醚= 1∶4),得到白色固體144mg。產率85%,θmp:263~265℃。化合物 4 的光譜數據為:IR(KBr)(υ/cm-1):3473,2921,2856,1732,1642,1445,1381,1238,1054;1H NMR(CDCl3,300MHz):4.404(1H,dd,J=14.7,7.5Hz,C16-H),3.34~3.51(2H,m,C26-H),0.975(1H,d,J=8.1Hz,21-CH3),0.930(1H,d,J=8.1Hz,27-CH3)0.790(6H,s,18-CH3and 19-CH3);13C NMR(CDCl3,75MHz) δ:165.7(3-C),109.3(22-C),80.8(16-C),66.8(26-C),62.2(17-C),56.19(14-C),54.1(9-C),41.6(5-C),40.6(4-C),40.5(20-C),40.0(13-C),38.9(12-C),38.0(2-C),36.2(1-C),35.0(10-C),32.0(8-C),31.75(15-C),31.4(23-C),30.7(7-C),30.3(24-C),29.7(6-C),28.8(25-C),21.0(11-C),17.1(18-C),16.5(27-C),14.5(19-C),11.5(21-C)。
1.3.5 3- 縮氨硫脲基劍麻皂素(5)的制備
將 204mg(0.492mmol) 3- 羰 基 劍 麻 皂 素 溶于20mL無水乙醇中,滴入冰醋酸調節溶液pH值為3~5,保持反應溫度為60℃,20min內分批加入54mg(0.261mmol)氨基硫脲,有白色固體析出。繼續反應10min,TLC監測(V乙酸乙酯∶V石油醚=1∶4),沒有原料點后停止反應,自然冷卻。減壓蒸去大部分溶劑,加入少量蒸餾水,用乙酸乙酯萃取,減壓蒸干得到白色固體。硅膠拌樣柱層析分離,先使用洗脫劑(V乙酸乙酯∶V石油醚=1∶4)將原料分離出來,再換用洗脫劑(V甲醇∶ V二氯甲烷=1∶20)將產品洗脫,得到白色固體214mg, 產率91.5%,θmp:270~271℃。化合物5的光譜數據:IR(KBr)(υ/cm-1):3456,3195,2929,2847,1683,1568,1454,1380,1246,1176,1066;1H NMR(CDCl3,300MHz)δ:8.762(1H,br s,-NH2),7.23(1H,s,-NH2),6.358(1H,s,-NH),4.38~4.41(1H,m,C16-H),3.33~3.49(2H,m,C26-H),0.968(3H,d,J=8.1Hz,21-CH3),0.928(3H,s,27-CH3),0.783(6H,s,18-CH3and 19-CH3);13C NMR(CDCl3,75MHz) δ:178.7(C=S),156.6(3-C),109.2(22-C),80.7(16-C),66.8(26-C),56.06(14-C),53.7(9-C),46.6(5-C),45.6(4-C),41.6(20-C),40.5(13-C),39.9(12-C),38.5(2-C),37.6(1-C),36.2(10-C),35.0(8-C),31.8(15-C),31.7(23-C),31.3(7-C),30.3(24-C),28.8(6-C),28.5(25-C),21.2(11-C),17.1(18-C),16.5(27-C),14.5(19-C),11.5(21-C)。
2 結果與討論
2.1 產物的譜圖分析
從化合物2的譜圖看,羥基峰消失,在1732cm-1處出現了羰基峰,這就說明劍麻皂素3- 位羥基已被氧化生成了羰基,也就是生成3- 羰基劍麻皂素(2)。
化合物3的紅外譜圖顯示在3383cm-1有肟羥基吸收峰,在1552cm-1出現了碳氮雙鍵的振動吸收峰,說明3- 位的羰基已被取代形成了肟羥基。在化合物3的1H NMR中在8.29×10-6處有一共振信號為肟羥基,這樣我們可以認為肟基已經存在于甾核,另外結合化合物3的13C NMR中出現161×10-6C=N官能團中C的吸收,進一步說明3- 位羰基被肟化。
與化合物2的譜圖相比,化合物4的紅外圖譜中出現氨基的特征峰,1642cm-1為碳氮雙鍵的吸收,再結合化合物4的13C NMR 中165.23×10-6出現3-位碳的碳氮雙鍵吸收,說明3- 位羰基已成功被腙基取代。
化合物5的紅外圖譜中1589cm-1為碳氮雙鍵的吸收,1503cm-1為碳硫雙鍵的吸收,1292cm-1為碳硫鍵的吸收。化合物5的1H NMR中低場出現了-NH2信號,6.39×10-6為-NH的共振信號, 說明3-位羰基已經轉變成為縮氨硫脲基。另外化合物5的13C NMR 中,原來化合物2的3- 位羰基碳的化學位移為210×10-6,在化合物5中已經向高場位移至156.6×10-6,178.7×10-6為碳硫雙鍵中C的化學位移,這說明化合物2的3- 羰基已經轉變成為3- 縮氨硫脲基,生成了3- 縮氨硫脲劍麻皂素(5)。
2.2 實驗結果與討論
在制備3- 羰基劍麻皂素(2)時,所加的原料與Collins試劑的量比例不同(1∶4、1∶5、1∶6),產率也不同,結果如表1所示。當劍麻皂素與Collins試劑的物質的量之比是1∶4,且在室溫條件下反應時,產率最高;同時反應也會受到溫度的影響,比較室溫和在35℃下的反應產率,可以知道升高溫度產率有所提高。
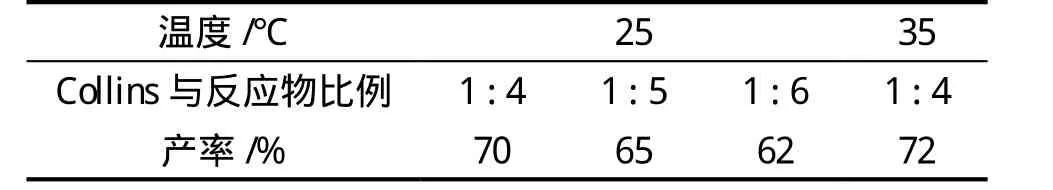
表1 Collins試劑用量與反應物產率
在制備3- 肟基劍麻皂素時(3)時,實驗過程中發現3- 羰基劍麻皂素(2)在95%乙醇中溶解性很好,而且反應中加入了CH3COONa·3H2O調節溶液的pH值為堿性,提高了羰基的親和性,使反應變得更容易,所以反應的產率很高,達到了90%以上。
3 結論
本文合成了3個新的不同的3- 取代劍麻皂素化合物,產率85%以上,合成方法簡單可行。合成產物的生理活性研究有待于進一步進行。
[1] 周寅,張夢宇,白楊,賈桂云.劍麻皂素的分離純化工藝研究[J].安徽農業科學,2011,39(8):4540-4542.
[2] 李燕婧,周桂芬,韋善新,鐘正賢.劍麻皂素藥理作用的研究[J].時珍國醫國藥,2006,17(10):1958-1959.
[3] 賴克道,李燕婧,李茂.劍麻皂素降血糖作用的研究[J].廣西科學院學報,2010,26(1):56-58.
[4] 韓廣甸, 馬兆揚.我國利用劍麻皂素合成甾體藥物的研究進展[J].中國醫藥工業雜志,2002,33(9):459-464.
[5] 韓耀玲,張東娣,林翠梧.劍麻皂素的硫酸酯化的研究[J].河南大學學報(醫學版),2007,26(4):31-33.
[6] Cui J. G. ,Huang L L,Fan L,Zhou A. M. A facile and efficient synthesis of some(6E)-hydroximino-4-en-3-one steroids,steroidal oximes from Cinachyrella spp. Sponges [J].Steroids, 2008,73 (3):252-256.
[1] Cui J. G.,Fan L,Huang L.,Liu H. Li,Zhou A. M.Synthesis and evaluation of some steroidal oximes as cytotoxic agents:Structure/activity studies(Ⅰ) [J].Steroids,2009,74(1):62-72.
