凝膠滲透色譜-氣相色譜-質譜聯用快速測定大豆中克百威、乙草胺、甲草胺、異丙甲草胺、氟樂靈的殘留量
2012-01-29 11:05:52蘇明明張旭東宋大賀
質譜學報 2012年1期
關鍵詞:大豆
蘇明明,張旭東,那 晗,王 琦,宋大賀,張 寧
(1.大連出入境檢驗檢疫局,遼寧 大連 116001;2.錦州出入境檢驗檢疫局,遼寧 錦州 121013;3.大窯灣出入境檢驗檢疫局,遼寧 大連 116001;4.遼寧出入境檢驗檢疫局,遼寧 大連 116100;5.大連機場檢驗檢疫局,遼寧 大連 116100)
大豆是重要的油料和蛋白來源,是我國大宗進口的農產品之一。克百威具有高活性、高效、低毒及易分解的特性,在世界范圍內被廣泛用于保護農作物免受蟲害。克百威是氨基甲酸酯類農藥,在高溫中具有熱不穩定性,在氣相色譜分析過程中,需同時測定克百威本體和其分解產物3-羥基克百威。目前,相關文獻的報道較多,但其主要缺點在于操作復雜、靈敏度低等[1-4],采用GC/MS法不僅可以同時進行克百威的定量測定和陽性確證,還可以提高測定的靈敏度及精確度。由于豆田中雜草繁多,氟樂靈、甲草胺、異丙甲草胺和乙草胺是大豆田常用的4種除草劑,主要用于防除禾本科雜草和一些闊葉雜草,每畝的登記施用劑量均小于100g(有效成分)。我國規定大豆中異丙甲草胺的殘留限量為0.02 mg/kg,氟樂靈的殘留限量為0.05mg/kg,甲草胺和乙草胺的殘留限量皆為0.2mg/kg。由于大豆中油脂類物質含量較高,凈化困難,試樣需采用凝膠滲透色譜(GPC)法去除色素和油脂。凝膠滲透色譜作為一種新的凈化方法和技術,在國外已廣泛使用[5-6],在我國的運用也日漸增多[7-10]。本研究采用凝膠滲透色譜-氣相色譜-質譜(GPC-GC/MS)法同時檢測大豆中氟樂靈、克百威、異丙甲草胺、甲草胺和乙草胺的殘留量,去除了油脂類物質的干擾,同時進行定量測定和陽性確證,希望能為大豆中5種農藥殘留的檢測提供新的方法。
1 試驗部分
1.1 儀器與試劑
6890-5975C氣相色譜-質譜儀:美國Agilent公司產品,配有7683B自動進樣器和分流/不分流進樣口;EI源及數據處理系統;Ultratur-Rax均質器,旋轉蒸發儀:德國IKA公司產品;離心機 RJ-TDL-40B:中國瑞江公司產品;移液槍(100~1 000μL):美國 Thermo公司產品;J2 AccuPrepMPS凝膠凈化色譜儀:美國J2公司產品。
克百威、乙草胺、甲草胺、異丙甲草胺和氟樂靈(質量濃度均為100mg/L):購自國家環境保護監測研究中心;流動相為V(環己烷)∶V(乙酸乙酯)=1∶1的溶液;正己烷、乙腈、乙酸乙酯和環己烷均為分析純,經重蒸餾后使用。
1.2 氣相色譜-質譜條件
1.2.1 氣相色譜條件 色譜柱:HP-5MS石英毛細柱(30m×0.25mm×0.25μm);升溫程序:80℃保持1min,以15℃/min升至270℃,保持10min;載氣(高純氦氣)流速1.2mL/min;壓力2.4kPa;進樣口溫度270℃;進樣量1μL;不分流進樣。
1.2.2 質譜條件 電子轟擊(EI)離子源;電子能量70eV;離子源溫度150℃,四極桿溫度230℃,色譜-質譜接口溫度280℃;全掃描范圍m/z 50~450用于測定樣品凈化效果,選擇離子監測用于定性和定量測定。
用乙草胺等5種農藥的混合標準溶液,在上述GC/MS條件下用Scan方式先測得其總離子流圖,然后根據各農藥的保留時間和質譜圖,確定各待測化合物在SIM測定方式中的采集時間和監測離子,列于表1。
1.3 凝膠色譜(GPC)凈化
1.3.1 凝膠色譜條件 Bio Beads S-X3凈化柱:700mm×25mm;流動相:V(環己烷)∶V(乙酸乙酯)=1∶1的混合溶液;流速:5.0 mL/min;樣品定量:5.0mL;預淋洗體積:50 mL;洗脫體積:200mL;收集體積:50mL。
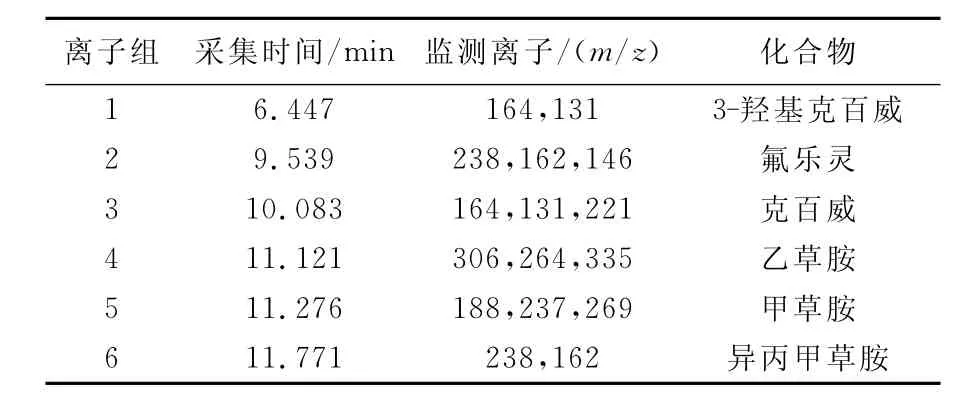
表1 GC/MS-SIM法分析5種農藥的采集時間和監測離子Table 1 End acquisition time and monitoring ion of GC/MS-SIM analysis for 5pesiticides
1.3.2 凝膠色譜凈化步驟 將10mL待凈化液按1.3.1進行凈化,合并餾分收集器中的收集液于250mL濃縮瓶中。
1.4 分析步驟
將大豆研碎,準確稱取5.0g粉碎樣品,加入50mL乙腈,均質2min,以4 000r/min離心10min,取乙腈層轉入100mL梨形瓶,再加入20mL乙腈重復上述提取1次,合并,于旋轉式蒸發儀中濃縮至干,加入10mL V(乙酸乙酯)∶V(環己烷)=1∶1的混合溶液定容,過0.45μm濾膜后,用GPC凈化。收集5~15min餾分,濃縮近干,用正己烷定容至5mL,待測。
2 結果與討論
2.1 GPC凈化方式
大豆及一些豆制品中含有大量的油脂和蛋白質等物質,當前最為常用的傳統經典提取方法有液-液分配法、層析法、凝結沉淀法、冷凍法、化學法、薄層色譜法、掃集共蒸餾法[11-13]等。如果樣品直接提取進樣難以除去大豆及其制品中的油脂,對色譜分析會產生一些干擾,影響定性定量分析的準確性。本實驗使用GPC進行凈化濃縮,通過對儀器條件的改進,實現了加壓和收集特定餾分的目的,同時也縮短了試樣凈化的時間。由于GPC是依靠被分離物質本身相對分子質量的大小對樣品進行分離的,凝膠本身與被分離試樣之間沒有相互作用,因此被分離物質在分離過程中不會發生化學變化。在GPC中,大分子物質將首先被洗脫出來,如油脂、類脂化合物、聚合物、蛋白質、色素及類固醇等,而小分子物質(如農藥等)則擴散進入凝膠孔內,較遲流出色譜柱。GPC的條件溫和,對農藥的性質沒有特殊要求,不同類型的農藥可同時分析,再加上在一個固定的淋洗系統中,凝膠可以被反復使用,所以利用Bio-Beads S-X3凝膠填料凈化樣本中的殘留農藥,不但技術成熟、廉價,而且重現性高、凈化和回收效果良好[10]。
2.2 監測離子的選擇
在SIM測定方式中,根據各化合物的質譜圖,選擇2~3個有足夠相對強度的離子作為選擇監測離子碎片用于定量測定和陽性確證。如果分子離子峰有足夠的相對強度(一般大于50%),則首選分子離子峰;其次是m/z>100的基峰或者是相對強度較大的其它離子碎片。
另外,克百威是氨基甲酸酯類農藥,在高溫中具有熱不穩定性,克百威有兩個峰出現,即克百威和其酚的代謝產物3-羥基克百威,所以克百威的測定結果為兩者之和,在SIM測定方式中設定3-羥基克百威的采集時間和監測離子[14]。GC/MS-SIM 測定方式下5種農藥的選擇離子流色譜圖示于圖1。
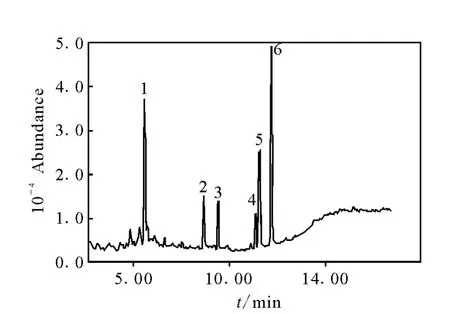
圖1 GC/MS-SIM測定方式下5種農藥(0.1mg/L)的總離子流圖Fig.1 GC/MS-SIM ion monitoring chromatograms of 5pesticides
2.3 標準工作曲線的繪制
根據各農藥的檢測靈敏度,取1.1節中的農藥標準儲備液,用正己烷逐級稀釋,配制成5種農藥的混合標準工作曲線。其中,克百威的濃度為0.005、0.01、0.025、0.1、0.2mg/L,甲草胺的濃度為0.01、0.025、0.05、0.1、0.2mg/L,乙草胺的濃度為0.01、0.02、0.05、0.1、0.2mg/L,異丙甲草胺的濃度為0.01、0.02、0.05、0.1、0.2 mg/L,氟樂靈的濃度為0.01、0.02、0.05、0.1、0.2mg/L,在1.2節的 GC/MS-SIM 條件進行測定。結果表明,各組分的濃度與峰面積的線性關系良好,相關系數為0.996 0~0.999 8,列于表2。

表2 標準曲線Table 2 Standard curve of detection
2.4 方法的回收率和精密度
將大豆樣品粉碎后,在大豆粉末中添加克百威、氟樂靈、甲草胺、異丙甲草胺和乙草胺進行回收率試驗,每個添加濃度重復6次,得出農藥回收率為66.26%~104.9%,相對標準偏差為2.2%~12.3%。本試驗在標準添加的最低質量濃度為0.01mg/L,5種農藥的信噪比均大于10,并有較好的回收率,故確定各農藥定量限為0.01mg/L,結果列于表3。
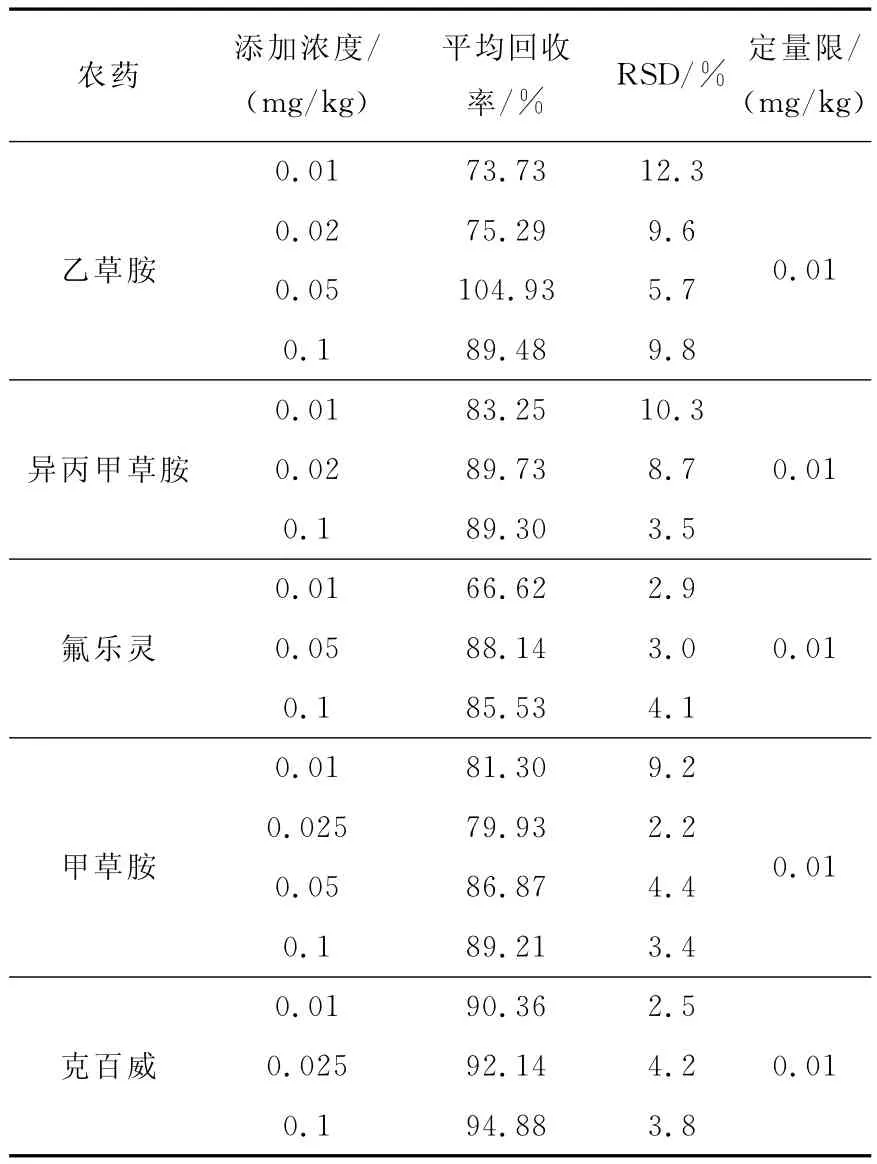
表3 5種農藥樣品的加標回收率及精密度Table 3 Recoveries and precisions of 5pesticides at different spiked concentrations
2.5 實樣檢測
自2010年4月以來,采用本方法檢測了大量的大豆樣品。對其中一批陽性樣品進行5次重復測定,結果為乙草胺,平均值為0.03 mg/kg,相對標準偏差為3.7%,其對應的色譜圖示于圖2~4。
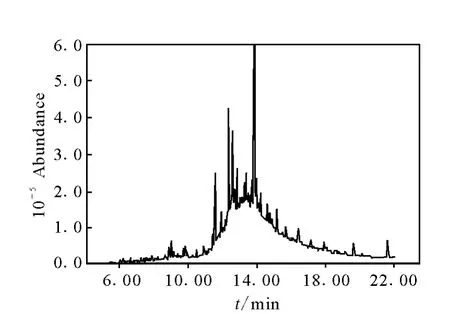
圖2 空白大豆樣品總離子色譜圖Fig.2 SIM chromatograms of soybean sample
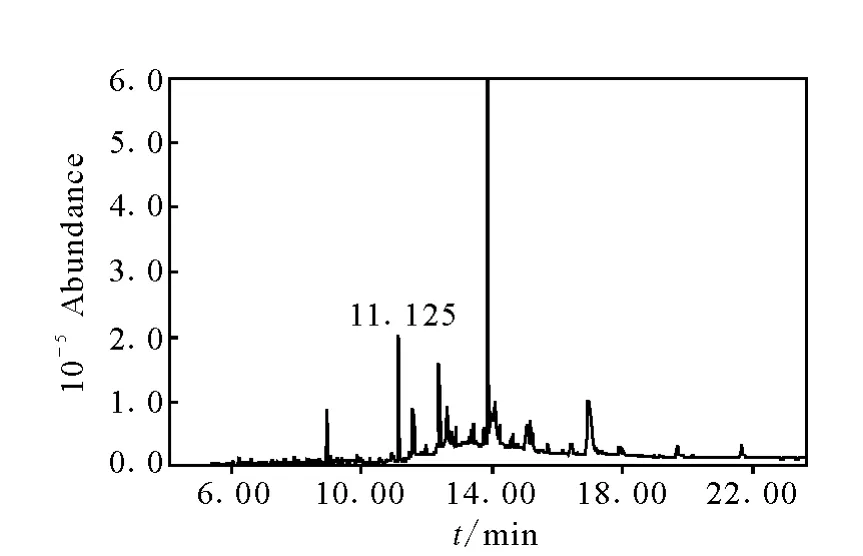
圖3 陽性大豆樣品總離子色譜圖Fig.3 SIM chromatograms of positive soybean sample
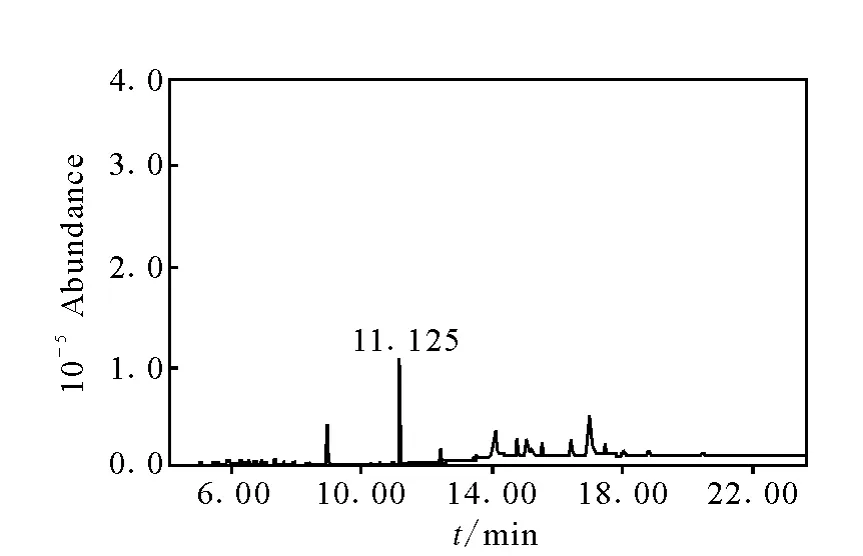
圖4 0.1mg/L乙草胺標準溶液的總離子色譜圖Fig.4 SIM chromatograms of 0.1mg/L acetochlor
3 結論
雖然檢測大豆中克百威、異丙甲草胺、氟樂靈、甲草胺和乙草胺的農藥殘留已有國家標準和其他檢測方法[15-18],但需要對5種農藥分別進行檢測,花費時間比較長。GPC結合GC/MS-SIM法可以對大豆中5種農藥殘留同時進行定量測定和陽性確證,達到了一舉兩得,事半功倍的效果,避免了氣相色譜法中當干擾物的保留時間與被測物非常接近時而引起的陽性判斷錯誤。GC/MS法中的SIM測定方式基線噪聲小、干擾少、具有與GC-FPD或GC-NPD法相當或者更高的靈敏度;測定克百威、異丙甲草胺、氟樂靈、甲草胺和乙草胺5種農藥殘留的檢出限量完全可以滿足檢測要求。
試樣用乙腈提取,GPC進行前處理,回收率、精密度較好、簡捷、快速,適用于在短時間內大批量樣品的分析。
[1]劉長武,劉瀟威,翟廣書,等.固相萃取-高效液相色譜法測定蔬菜水果中的氨基甲酸酯殺蟲劑及其代謝物殘留[J].色譜,2003,21(3):255-257.
[2]賀小雄,郭 萍,趙鳳英,等.底泥中多種有機氯及擬除蟲菊酯類農藥殘留同時測定法[J].江西理工大學學報,2008,29(3):30-33.
[3]李 英,周艷明,牛 森.蔬菜、水果中氨基甲酸酯類農藥多殘留分析方法的研究[J].現代科學儀器,2005,(6):68-70.
[4]陳曉紅,仇佩紅,金米聰.水樣中氨基甲酸酯農藥及其代謝產物的分析方法研究[J].中國衛生檢驗雜志,2006,16(1):9.
[5]TOMOMI G,YUKO I.Simple and rapid determination of Nmethylcarbamate pesticides incitrus fruits by electro spray ionization tandem mass spectrometry[J].Analytica Chimica Acta,2003,487(2):201-209.
[6]ERIC J,JOSEP M B.Trace level determination of organochlorine,organophosphorus and pyrethroid pesticides in lanolin using gel permeation chromatography followed by dual gas chromatography and gas chromatography-negative chemical ionization mass spectrometric confirmation[J].Journal of chromatography A,2002,950(1/2):213-220.
[7]魏冬旭,江連洲,郭 偉,等.ASE-GPC-GC法測定大豆及豆制品中六六六、滴滴涕農藥殘留[J].食品科學,2009,30(24):351-354.
[8]李 櫻,儲曉剛,仲維科,等.凝膠滲透色譜和固相萃取凈化2氣相色譜分離組合法測定糙米中的殘留農藥[J].分析化學,2004,32(10):1 325-1 328.
[9]張偉國,儲曉剛,李重九.凝膠滲透色譜技術用于檢測大米中擬除蟲菊酯類農藥的殘留量[J].農藥,2005,44(8):372-373.
[10]紀欣欣,石志紅,曹彥忠,等.凝膠滲透色譜凈化/液相色譜-串接質譜法對動物脂肪中111種農藥殘留量的同時測定[J].分析測試學報,2009,28(12):1 433-1 439.
[11]陳 躍,盧曉宇,趙汗青,等.氣相色譜質譜聯用法對農產品中8種衣劑農藥殘留的同時測定[J].分析測試學報,2008,27:1 084-1 087.
[12]靳保輝,陳沛金,謝麗琪,等.茶葉中25種農藥多殘留氣相色譜測定方法[J].分析測試學報,2007,26(1):104-106.
[13]賀小雨,陳樹兵,俞雪鈞,等.冷凍去脂-固相萃取/氣相色譜-質譜法對水產品中的禾草枯、溴氰菊酯及19種有機氯農藥殘留的測定[J].分析測試學報,2009,28(3):306-309.
[14]王 建,林秋萍,雷鄭莉,等.氣相色譜-質譜法測定蔬菜中有機磷殺蟲劑和克百威的殘留量[J].分析實驗室,2002,21(2):282-284.
[15]潘燦平,王麗敏,孔祥雨,等.凝膠色譜凈化-毛細管氣相色譜測定黃瓜、番茄和青椒中15種有機磷農藥[J].色譜,2002,20(6):565-568.
[16]張偉國,儲曉剛,李重九.氣相色譜/離子阱質譜-選擇離子方法同時檢測大米中百種農藥殘留[J].分析化學,2006,34(4):484-488.
[17]藏小丹,牟光慶.氣相色譜法同時測定大豆中多種除草劑的殘留[J].食品研究與開發,2010,31(3):132-133.
[18]胡 敏,李二虎,吳兵兵,等.色譜-質譜聯用檢測茶葉中多種殘留農藥的研究[J].現代農藥,2006,45(6):121.
猜你喜歡
農業科技通訊(2023年1期)2023-02-12 07:09:18
今日農業(2022年16期)2022-11-09 23:18:44
中國化肥信息(2022年7期)2022-08-31 01:29:28
中國化肥信息(2022年5期)2022-08-30 01:58:26
今日農業(2021年13期)2021-11-26 11:50:54
今日農業(2021年20期)2021-11-26 01:23:56
今日農業(2021年14期)2021-10-14 08:35:34
下一代英才(酷炫少年)(2018年6期)2018-07-09 03:17:44
農產品市場周刊(2017年4期)2017-03-03 19:40:05
兒童故事畫報·智力大王(2015年10期)2016-01-27 01:01:35
