中國制藥企業(yè)原料藥出口的國際認(rèn)證
2011-11-11 07:54:12權(quán)秀麗
中國醫(yī)藥導(dǎo)報(bào) 2011年23期
馮 煜,權(quán)秀麗,崔 洋
步長集團(tuán)生產(chǎn)運(yùn)營管理中心,陜西西安 710075
中國制藥企業(yè)原料藥出口的國際認(rèn)證
馮 煜,權(quán)秀麗,崔 洋
步長集團(tuán)生產(chǎn)運(yùn)營管理中心,陜西西安 710075
本文具體詳實(shí)地描述了中國制藥企業(yè)原料藥出口歐洲和美國等發(fā)達(dá)市場時(shí)藥品注冊(cè)所應(yīng)具備的基本條件,明確地描述了藥物原料藥注冊(cè)時(shí)申請(qǐng)F(tuán)DA認(rèn)證,歐盟EDMF申請(qǐng)/COS證書認(rèn)證所需要的文件、文件的格式以及認(rèn)證的詳細(xì)程序等,并且對(duì)未來全球范圍內(nèi)在ICH指南的前提下統(tǒng)一和規(guī)范藥品注冊(cè)所使用的CTD格式文件作了明確的說明,目的在于使中國原料藥出口企業(yè)對(duì)于國際認(rèn)證的現(xiàn)狀和未來有所了解,明確認(rèn)識(shí)國際注冊(cè)認(rèn)證程序和所需要的文件,生產(chǎn)工廠所應(yīng)具備的基本條件等,并能對(duì)加快我國原料藥的出口起到一定的促進(jìn)作用。
原料藥;藥物注冊(cè);FDA 認(rèn)證;DMF;EDMF;COS 認(rèn)證
眾所周知,在過去的10年里越來越多的中國生產(chǎn)廠家都在尋求各種各樣的出口機(jī)會(huì),同時(shí)也面臨了越來越多的國外政府機(jī)構(gòu)或者制劑企業(yè)嚴(yán)格的質(zhì)量審計(jì),中國的制藥企業(yè)也借此機(jī)會(huì)不斷提高了自身的質(zhì)量管理體系,安全管理體系以及整體企業(yè)形象。
原料藥企業(yè)產(chǎn)品出口面向的主要地區(qū)為美國和歐盟國家,理所當(dāng)然原料藥出口企業(yè)必須通過上述國家和地區(qū)質(zhì)量,生產(chǎn)體系的審計(jì)(GMP),并完成產(chǎn)品注冊(cè)。產(chǎn)品注冊(cè)涉及到美國食品及藥物管理局(Food and Drug Admistraton,F(xiàn)DA)認(rèn)證和歐盟不同國家藥物主文件(drug master file,DMF)注冊(cè),或者歐盟整體的歐州藥典適用性證書(certificate of suitabitity,COS)認(rèn)證。
1 FDA認(rèn)證介紹
根據(jù)美國的聯(lián)邦管理法規(guī)定,藥品進(jìn)入美國須向美國FDA申請(qǐng)注冊(cè)并遞交有關(guān)文件,化學(xué)原料藥按要求提交一份DMF。
1.1 DMF文件及申報(bào)程序簡介
DMF是由生產(chǎn)商提供的某藥品生產(chǎn)全過程的詳細(xì)資料,便于FDA對(duì)該廠產(chǎn)品進(jìn)行全面了解,內(nèi)容包括:生產(chǎn)、加工、包裝和貯存某一藥物時(shí)所用的具體廠房設(shè)施和監(jiān)控的資料,以確定藥品的生產(chǎn)是在GMP環(huán)境下進(jìn)行的[1]。
1.1.1 DMF文件共有五種類型 Ⅰ型:生產(chǎn)地點(diǎn)和廠房設(shè)施;Ⅱ型:中間體、原料藥和藥品;Ⅲ型:包裝物料;Ⅳ型:輔料、著色劑、香料、香精及其他添加劑;Ⅴ型:非臨床數(shù)據(jù)資料和臨床數(shù)據(jù)資料。原料藥生產(chǎn)企業(yè)向FDA申報(bào)的DMF文件屬于Ⅱ型,申請(qǐng)文件的主要內(nèi)容包括遞交申請(qǐng)書、相關(guān)行政管理信息、企業(yè)的承諾聲明、申請(qǐng)產(chǎn)品的物理和化學(xué)性質(zhì)描述、產(chǎn)品生產(chǎn)方法詳述、產(chǎn)品質(zhì)量控制與生產(chǎn)過程控制、產(chǎn)品穩(wěn)定性實(shí)驗(yàn)、包裝和標(biāo)簽、標(biāo)準(zhǔn)操作規(guī)程、原材料及成品的貯存與管理、文件管理、驗(yàn)證、批號(hào)管理制度、退貨及處理。
1.1.2 DMF文件主要內(nèi)容包括如下幾個(gè)方面 ①綜述資料:A.行政信息;B.產(chǎn)品特征的綜述;C.專家報(bào)告。②化學(xué)、藥學(xué)和生物學(xué)信息:A.配方的詳細(xì)內(nèi)容;B.生產(chǎn)方法的描述;C.起始物料的描述;D.特殊檢測(cè);E.生產(chǎn)中間過程的控制;F.最終產(chǎn)品檢驗(yàn);G.穩(wěn)定性試驗(yàn)。③生物等效性試驗(yàn)結(jié)果:對(duì)于大部分藥品,生物等效性試驗(yàn)數(shù)據(jù)是必須的。
1.1.3 DMF申報(bào)的基本程序 見圖1。
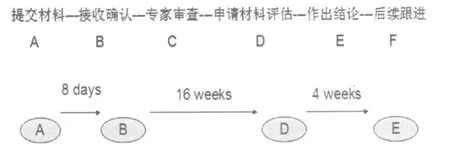
圖1 DMF申報(bào)的基本程序
1.2一般情況下中國廠商申報(bào)FDA認(rèn)證的程序如下[2]
1.2.1 進(jìn)行國際市場調(diào)研,摸清美國市場目前的銷售情況,對(duì)市場發(fā)展趨勢(shì)與走向做出正確的預(yù)測(cè)、分析和判斷,選擇好申請(qǐng)F(tuán)DA批準(zhǔn)的品種。
1.2.2 選擇申請(qǐng)代理人和代理經(jīng)銷商,并簽訂委托協(xié)議書、簽署委托書。
1.2.3 編寫申請(qǐng)文件,原料藥為DMF文件,由代理人完成申請(qǐng)文件終稿的編寫并向FDA遞交,取得DMF文件登記號(hào)。
1.2.4 FDA收到申請(qǐng)文件后,經(jīng)初審合格后發(fā)通知函給申請(qǐng)人,并發(fā)給一個(gè)登記號(hào),說明DMF文件持有人的責(zé)任和義務(wù)。
1.2.5 工廠按美國cGMP的要求進(jìn)行廠房、設(shè)施設(shè)備的改造并完善生產(chǎn)質(zhì)量管理的各項(xiàng)軟件和相關(guān)人員的強(qiáng)化培訓(xùn)。
1.2.6 應(yīng)美國制劑生產(chǎn)廠家(即該原料藥品的終端用戶)的申請(qǐng),F(xiàn)DA派官員到生產(chǎn)廠家按照FDA頒布的生產(chǎn)現(xiàn)場檢查指南并對(duì)照已上報(bào)審核的DMF文件進(jìn)行檢查,F(xiàn)DA官員在生產(chǎn)現(xiàn)場的基礎(chǔ)上出具書面意見給生產(chǎn)廠家并向FDA報(bào)告檢查結(jié)果。
1.2.7 FDA審核批準(zhǔn)后將審核結(jié)果通知生產(chǎn)廠家并輸入美國海關(guān)的管理系統(tǒng),該原料藥品即獲準(zhǔn)直接進(jìn)入美國市場。
1.2.8 生產(chǎn)廠家每年向FDA遞交一份DMF修改材料,一般情況下,每2~3年可能要接受一次復(fù)查。
1.3 FDA的批準(zhǔn)
按照美國聯(lián)邦法規(guī)第210及第211條中有關(guān)規(guī)定,任何進(jìn)入美國市場的藥品(包括原料藥品)都需要首先獲得FDA的批準(zhǔn),而且所有關(guān)于藥物的生產(chǎn)、加工、包裝等均應(yīng)嚴(yán)格符合美國cGMP的要求。
1.3.1 原料藥通過美國FDA認(rèn)證的途徑 對(duì)于原料藥來說,通過FDA批準(zhǔn)主要有兩個(gè)階段:一是DMF文件的登記,要求遞交的DMF文件對(duì)所申請(qǐng)的藥品的生產(chǎn)和質(zhì)量管理的全過程以及藥品質(zhì)量本身做一個(gè)詳盡的描述。二是當(dāng)DMF文件的登記已完成時(shí),而且在美國的原料藥品終端用戶提出了申請(qǐng)以后,F(xiàn)DA官員對(duì)原料藥物的生產(chǎn)廠家進(jìn)行GMP符合性現(xiàn)場檢查,通過對(duì)藥品生產(chǎn)全過程的生產(chǎn)管理和質(zhì)量管理狀況的全面考察,做出該原料藥生產(chǎn)企業(yè)的生產(chǎn)和質(zhì)量管理能否確保所生產(chǎn)藥品的質(zhì)量的判斷。
1.3.2 DMF文件的編寫和提交 DMF編寫應(yīng)實(shí)事求是,因?yàn)镕DA現(xiàn)場檢查會(huì)以企業(yè)提交的DMF作為檢查的依據(jù)。DMF必須用英文書寫,如果呈報(bào)文件是以其他語言書寫的,就必須附有正確的英文譯文,連同授權(quán)信、副本一同交給美國代理商,由代理商交給FDA。FDA審閱DMF之后,若認(rèn)為符合模式要求,則會(huì)給該DMF一個(gè)注冊(cè)號(hào),同時(shí)將該注冊(cè)號(hào)通知美國代理商,表明FDA已經(jīng)收到了符合模式要求的DMF。
1.3.3 FDA的現(xiàn)場檢查 FDA現(xiàn)場檢查一般是非無菌原料藥由2個(gè)人檢查 3~4 d,無菌原料藥由2~3個(gè)人檢查 5~7 d。現(xiàn)場檢查有兩種形式:一是從頭開始,按操作順序的先后進(jìn)行檢查,這種按產(chǎn)品生產(chǎn)流程的檢查一般適用于一種產(chǎn)品;另一種是按系統(tǒng)檢查,適用于多個(gè)品種的檢查。現(xiàn)場檢查時(shí)FDA會(huì)提出大量問題,并查看企業(yè)的文件和記錄,最后FDA會(huì)有半天時(shí)間進(jìn)行總結(jié),若有疑問或認(rèn)為有些地方不符合GMP和DMF,他們會(huì)在審計(jì)缺陷483表上列出。通常483表上的問題要被審查企業(yè)在3周內(nèi)列出詳細(xì)的解釋或整改計(jì)劃并交FDA。當(dāng)FDA對(duì)企業(yè)的483表回信進(jìn)行審查,并感到滿意時(shí),F(xiàn)DA會(huì)通知代理商,同意其使用本產(chǎn)品。同時(shí)FDA會(huì)通知美國的貿(mào)易部門,準(zhǔn)許進(jìn)口本原料藥(FDA不會(huì)給企業(yè)發(fā)GMP證書)。經(jīng)過以上幾個(gè)步驟,企業(yè)的原料藥已經(jīng)成功進(jìn)入美國市場,但需要指出的是,并不是通過檢查之后就萬事大吉了,企業(yè)從此更應(yīng)嚴(yán)格按照GMP和DMF的要求運(yùn)作,重大變更應(yīng)通知FDA,DMF及時(shí)更新,同時(shí)FDA會(huì)至少每2年對(duì)企業(yè)復(fù)查一次[3]。
2 歐洲國家藥物注冊(cè)管理
2.1 EDMF文件簡介
歐洲藥物主文件(European drug master file,EDMF)是藥品制劑的制造商為取得上市許可而必須向注冊(cè)當(dāng)局提交的關(guān)于在制劑產(chǎn)品中所使用原料藥的基本情況的支持性技術(shù)文件。它的申請(qǐng)必須與使用該原料藥制劑的上市許可申請(qǐng)同時(shí)進(jìn)行。
EDMF分為公開部分和保密部分。與美國FDA的DMF涵蓋藥品生產(chǎn)的全過程CMC(chemistry,manufacturing and control)不同,EDMF的主要內(nèi)容是藥物及其相關(guān)雜質(zhì)的化學(xué),包括化學(xué)結(jié)構(gòu)及結(jié)構(gòu)解析、化學(xué)性質(zhì)、雜質(zhì)及其限度、雜質(zhì)檢查等。
2.2 EDMF的適用范圍
EDMF適用于以下三類原料藥的申請(qǐng):①仍由專利保護(hù)的新的原料藥,并且這種原料藥沒有包括在歐洲或任何一個(gè)成員國的藥典中;②已過專利保護(hù)期的原料藥,并且這種原料藥沒有包括在歐洲或任何一個(gè)成員國的藥典中;③包括在歐洲或任何一個(gè)成員國的藥典中的原料藥。
2.3 EDMF的變動(dòng)和更新
如果EDMF持有人需要對(duì)EDMF的公開部分和保密部分作出任何變動(dòng),均要向主管當(dāng)局或EMEA上報(bào),并通知所有申請(qǐng)人。若僅是修改EDMF的保密部分,并且生產(chǎn)采用的質(zhì)量標(biāo)準(zhǔn)和雜質(zhì)范圍均沒有發(fā)生改變,修改信息只需提供給主管當(dāng)局。如果需要修改EDMF的公開部分,此信息必須提供給其他申請(qǐng)人和使用此EDMF藥品上市許可證的持有人,所有涉及到的申請(qǐng)人將通過適當(dāng)?shù)淖兏绦蛐薷乃麄兊纳鲜性S可證申請(qǐng)文檔。
2.4 EDMF的遞交程序
根據(jù)歐洲藥物管理檔案程序的要求,EDMF只能在遞交制劑藥品上市許可證申請(qǐng)時(shí)遞交,并且只有歐洲的制劑生產(chǎn)廠家及其授權(quán)的代表(如進(jìn)口商)才能遞交EDMF。遞交的EDMF應(yīng)包括兩個(gè)部分:①EDMF的申請(qǐng)人部分(即公開部分);②原料藥生產(chǎn)廠家(ASM)的限制部分(即保密部分)。兩個(gè)部分要分開遞交,因?yàn)樗幤飞鲜性S可證的申請(qǐng)人是不可以看到EDMF保密部分的,其中EDMF的公開部分和保密部分組成的一個(gè)完整副本由原料藥生產(chǎn)廠家直接寄給歐洲的相關(guān)的評(píng)審機(jī)構(gòu)。公開部分的一個(gè)副本由原料藥生產(chǎn)廠家提前寄給申請(qǐng)人,并由申請(qǐng)人將此部分包括在上市許可證的申請(qǐng)文件中。
如果經(jīng)歐洲的藥品評(píng)審機(jī)構(gòu)驗(yàn)證,證明遞交的EDMF申請(qǐng)文件是真實(shí)有效的,則給予藥品上市許可證的申請(qǐng)人一個(gè)EDMF登記號(hào)。這樣作為原料藥的生產(chǎn)廠家,就可以將本原料藥產(chǎn)品出口到歐洲,用于該制劑廠家的藥品生產(chǎn)。
2.5 EDMF的評(píng)估
當(dāng)EDMF文件被提交后,歐洲各國的主管當(dāng)局或歐洲藥物評(píng)審局(EMEA,European agency for the evaluation of medicinal product)會(huì)對(duì)EDMF的公開部分和保密部分進(jìn)行評(píng)估并提問,且對(duì)EDMF公開部分的提問會(huì)寫進(jìn)整個(gè)評(píng)估報(bào)告并轉(zhuǎn)給申請(qǐng)人,對(duì)EDMF保密部分的提問則被包含在評(píng)估報(bào)告的保密附件內(nèi)直接轉(zhuǎn)給EDMF持有人,但主管當(dāng)局或EMEA會(huì)將上述情況連同所提問題的性質(zhì)通知申請(qǐng)人,申請(qǐng)人負(fù)責(zé)EDMF持有人應(yīng)及時(shí)解答這些問題。一旦因?yàn)檫@些針對(duì)保密部分的提問和解答使得公開部分內(nèi)容發(fā)生變動(dòng),EDMF持有人將有責(zé)任向申請(qǐng)人提供更新的公開部分的文件,并由申請(qǐng)人提供給評(píng)審機(jī)構(gòu)。
2.6 COS認(rèn)證介紹
歐洲藥典適用性證書(certificate of suitability,COS)認(rèn)證的目的是考察歐洲藥典是否能夠有效地控制進(jìn)口藥品的質(zhì)量,這是中國原料藥合法地被歐盟最終用戶使用的另一種注冊(cè)方式。這種注冊(cè)途徑的優(yōu)點(diǎn)是不依賴于最終用戶,可以由原料藥生產(chǎn)廠商獨(dú)立地提出申請(qǐng)。中國的原料藥生產(chǎn)廠商向歐盟藥品質(zhì)量指導(dǎo)委員會(huì)(EDQM)提交產(chǎn)品的COS認(rèn)證文件,在文件審查和可能的現(xiàn)場考察通過之后,EDQM會(huì)向原料藥品的生產(chǎn)廠商頒發(fā)COS認(rèn)證證書。
1999年,在EDQM制訂的COS認(rèn)證指南中提出:原料藥生產(chǎn)企業(yè)在COS認(rèn)證的申請(qǐng)文件中必須附加兩封承諾信,一封信承諾其申報(bào)的原料藥是按照國際GMP規(guī)范(ICH Q7A)進(jìn)行生產(chǎn)的,另一封信要求承諾同意歐洲GMP檢查機(jī)構(gòu)的官員進(jìn)行現(xiàn)場檢查。
隨著美國、歐盟和日本三方在藥品注冊(cè)程序和法規(guī)上的相互協(xié)調(diào),歐盟在進(jìn)口的原料藥注冊(cè)中逐步接近美國FDA的偏重現(xiàn)場GMP檢查的辦法,今后有可能對(duì)每一家提出COS認(rèn)證的生產(chǎn)廠家進(jìn)行現(xiàn)場的GMP檢查。COS認(rèn)證過程對(duì)企業(yè)是有積極意義的,會(huì)使企業(yè)的GMP管理達(dá)到國際水平,而且隨著美、歐、日三方協(xié)調(diào)的進(jìn)一步發(fā)展,通過歐盟的GMP檢查和COS認(rèn)證最終有可能直接進(jìn)入美國和日本市場,至少會(huì)使美國FDA的注冊(cè)變得更為容易。
2.7 EDMF申請(qǐng)和COS證書的比較
EDMF和COS證書都是原料藥進(jìn)入歐洲市場有效而必需的支持性材料,二者都是用于證明制劑產(chǎn)品中所使用的原料藥質(zhì)量的文件,以便支持使用該原料藥的制劑產(chǎn)品在歐洲的上市申請(qǐng)。
2.7.1 評(píng)審方式上的不同 EDMF是由單個(gè)國家的機(jī)構(gòu)評(píng)審的,是作為制劑上市許可申請(qǐng)文件的一部分,與整個(gè)制劑上市許可的申請(qǐng)文件一起進(jìn)行評(píng)審。無論原料藥物用于哪個(gè)制劑的生產(chǎn),或該EDMF是否已進(jìn)行過登記,都要進(jìn)行重新評(píng)審,因此對(duì)原料藥的生產(chǎn)廠家來說,不同的廠家要多次申請(qǐng)登記,可能花費(fèi)更多的時(shí)間和精力。而COS申請(qǐng)文件是由有關(guān)當(dāng)局組成的專家委員會(huì)集中評(píng)審的,評(píng)審結(jié)果將決定是否頒發(fā)COS證書。一個(gè)原料藥一旦取得COS證書,就可以用于歐洲藥典委員會(huì)的31個(gè)成員國內(nèi)所有制劑生產(chǎn)廠家的制劑生產(chǎn)。
2.7.2 針對(duì)的情況不同 EDMF與使用該原料藥的制劑藥物的上市許可申請(qǐng)不可分離,必須由使用該原料藥的歐洲終端用戶申請(qǐng);而COS證書則是直接將證書頒發(fā)給原料藥的生產(chǎn)廠家,因此它可由原料藥生產(chǎn)廠家獨(dú)立申請(qǐng),并不需要現(xiàn)成的中間商和終端用戶,因而生產(chǎn)廠家在申請(qǐng)過程中更加主動(dòng)。
2.7.3 適用的范圍不同 EDMF程序適用于所有的原料藥品,只要是原料藥,無論是否已收載入歐洲藥典,都可以通過EDMF文件的方式進(jìn)入歐洲市場,而COS證書只能處理歐洲藥典已收載的藥品,當(dāng)然不僅是原料藥,也包括生產(chǎn)制劑所用的輔料,我國的藥用輔料也可以申請(qǐng)COS證書。
2.7.4 所要求提供的資料不同 比如EDMF文件必須包括藥物的穩(wěn)定性研究資料,而COS證書的申請(qǐng)文件并不強(qiáng)求這些資料。
2.7.5 申請(qǐng)的結(jié)果不同 申請(qǐng)COS證書的結(jié)果是直接頒發(fā)給原料藥的生產(chǎn)廠家一個(gè)證書,只要將這個(gè)證書的復(fù)印件提供給歐洲方面的中間商或終端用戶,對(duì)方就可以購買本原料藥,而EDMF文件登記的結(jié)果只是告訴制劑生產(chǎn)廠家一個(gè)EDMF文件的登記號(hào),歐洲評(píng)審機(jī)構(gòu)不會(huì)將這個(gè)登記號(hào)告訴原料藥的生產(chǎn)廠家,原料藥的生產(chǎn)廠家只能從負(fù)責(zé)申請(qǐng)登記的歐洲藥品制劑的生產(chǎn)廠家查詢這個(gè)登記號(hào)。
3 CTD文件簡介
隨著由美國、歐洲和日本三方發(fā)起的國際協(xié)調(diào)會(huì)議(international conference of harmonization of technical requirements for registration of pharmaceuticals for human use,ICH)的進(jìn)程,在上述三個(gè)地區(qū)對(duì)于人用藥申請(qǐng)注冊(cè)的技術(shù)要求方面已經(jīng)取得了相當(dāng)大的協(xié)調(diào)統(tǒng)一,ICH決定采用統(tǒng)一的格式來規(guī)范各個(gè)地區(qū)的注冊(cè)申請(qǐng),并在2003年7月起首先在歐洲實(shí)行,這就是我們所說的CTD格式的注冊(cè)文件[4]。
3.1 CTD文件的組成及排版格式
3.1.1 CTD文件的組成 CTD文件是國際公認(rèn)的文件編寫格式,用來制作一個(gè)向藥品注冊(cè)機(jī)構(gòu)遞交的結(jié)構(gòu)完善的注冊(cè)申請(qǐng)文件,共由五個(gè)模塊組成。模塊一:行政信息和法規(guī)信息。本模塊包括各地區(qū)特殊的文件,例如申請(qǐng)表或在各地區(qū)被建議使用的標(biāo)簽,其內(nèi)容和格式可以由每個(gè)地區(qū)的相關(guān)注冊(cè)機(jī)構(gòu)來指定。模塊二:CTD文件概述[5]。本模塊是對(duì)藥物質(zhì)量、非臨床和臨床實(shí)驗(yàn)方面內(nèi)容的高度總結(jié)概括,必須由合格的、有經(jīng)驗(yàn)的專家來擔(dān)任文件編寫工作。模塊三:質(zhì)量部分。文件提供藥物在化學(xué)、制劑和生物學(xué)方面的內(nèi)容。模塊四:非臨床研究報(bào)告。文件提供原料藥和制劑在毒理學(xué)和藥理學(xué)試驗(yàn)方面的內(nèi)容。模塊五:臨床研究報(bào)告。文件提供制劑在臨床試驗(yàn)方面的內(nèi)容。其中,模塊一是地區(qū)特異性的,模塊二、三、四和五在各個(gè)地區(qū)是統(tǒng)一的。CTD文件組成的基本模塊,見圖2。
3.1.2 CTD文件寫作排版格式的要求 見圖3。
3.2 CTD文件實(shí)行的意義
在國際藥品注冊(cè)申請(qǐng)的技術(shù)文件編寫中采用統(tǒng)一的格式將會(huì)顯著減少企業(yè)財(cái)力和物力的投入,縮短申請(qǐng)編寫的時(shí)間,這些標(biāo)準(zhǔn)化的文件還將有助于注冊(cè)機(jī)構(gòu)的評(píng)審,并加強(qiáng)同申請(qǐng)人之間的交流。此外,在各注冊(cè)機(jī)構(gòu)之間注冊(cè)資料的交換也將隨之被簡化。通過CTD來提供資料,資料內(nèi)容將更加清晰透明,有利于文件中基礎(chǔ)數(shù)據(jù)的評(píng)審,幫助評(píng)審人快速定位所申請(qǐng)的內(nèi)容。
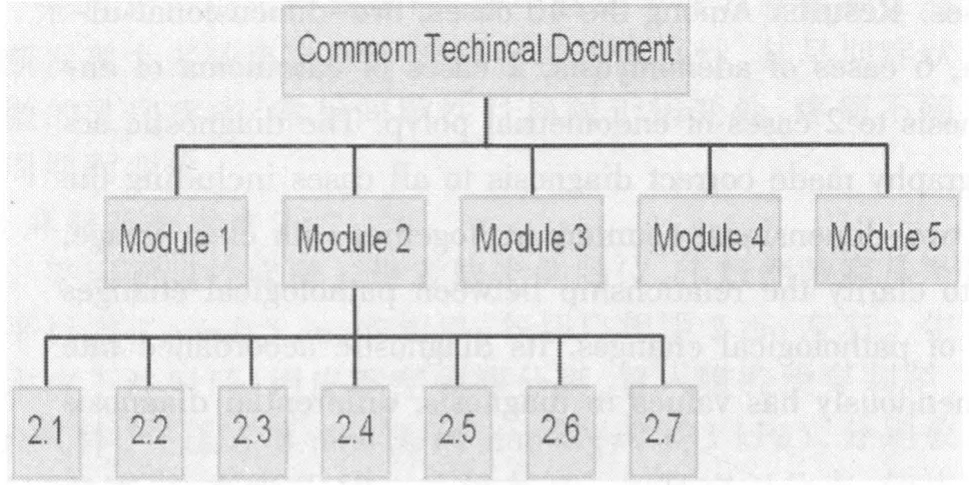
圖2 CTD文件組成的基本模塊

圖3 CTD文件寫作排版格式的要求
歐盟從2003年7月1日起強(qiáng)制性推行CTD格式。因此,歐盟藥品評(píng)價(jià)局(EMEA)規(guī)定2003年7月1日以后在歐盟申請(qǐng)注冊(cè)的EDMF必須使用CTD格式,在此之前向任何歐盟成員國提交的EDMF文件可以繼續(xù)用舊格式提交補(bǔ)充或修改文件,但2004年12月31日以后將不再接受任何舊格式的補(bǔ)充文件。
[1]US Food and Drug Administration.Guidance of compliance and regulatory information[OL].http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm122886.htm,2011-02-06.
[2]Food and Drug Administration.Guidance for Industry,Quality Systems Approach to Pharmaceutical CGMP Regulations[EB/OL].http://www.FDA.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070337.pdf,2010-09-22.
[3]Food and Drug Administration.Guidance for Industry,ANDAS:Impurities in Drug Substances[EB/OL].http://www.FDA.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM172002.pdf,2010-09-09.
[4]ICH.Process of Harmonization[OL].http://www.ich.org/about/process-ofharmonisation.html,2011-03-20.
[5]ICH.Q7A Good manufacturing practice[OL].http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q7/Step4/Q7_Guideline.pdf,2010-02-28.
International registration of API for Chinese pharmaceutical company
FENG Yu,QUAN Xiuli,CUI Yang
Production&Operations management center,Buchang Pharmaceuticals,Shaanxi710075,China
The article clearly describes the basic drug registration requirements for Chinese pharmaceutical company who exported APIs to US and European countries,and other developed markets,the registration dossier′s format,especially the procedures for FDA approval and getting EDMF/COS certificate were also introduced in detail,and the CTD format under ICH guideline which was used in unitying and regulating the drug registration were also explained definitely.The purpose was to make Chinese local pharmaceutical company to understand the international authentication of the present and future and know the authentication procedures international registration files needed clearly and the basic conditions of the production factory and so on,and be a certain role in pushing the speed in the export of the API.
Active pharmaceutical ingredients;Drug registration;Food and Drug Admistraton approval;Drug master file;European drug master file;Certificate of suitability;European drug master file;International drug registration
TQ460.4
C
1673-7210(2011)08(b)-111-04
馮煜(1971.3-),女,陜西人,大學(xué)本科,步長集團(tuán)生產(chǎn)運(yùn)營管理中心質(zhì)量經(jīng)理,主要從事醫(yī)藥企業(yè)質(zhì)量保證體系管理及CGMP符合性審計(jì)/法規(guī)事務(wù)等方面研究。
2011-04-11)
猜你喜歡
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
小學(xué)科學(xué)(學(xué)生版)(2020年10期)2020-10-28 07:52:12
中國化肥信息(2020年7期)2020-03-19 01:54:02
小學(xué)生優(yōu)秀作文(低年級(jí))(2018年6期)2018-05-19 01:54:28
中國軍轉(zhuǎn)民(2017年6期)2018-01-31 02:22:28
中國衛(wèi)生(2016年5期)2016-11-12 13:25:28
中國衛(wèi)生(2015年9期)2015-11-10 03:11:14
中國衛(wèi)生(2015年5期)2015-11-08 12:09:48
汽車零部件(2014年11期)2014-09-18 11:57:16
