PI3K/AKT通路在肺癌轉移和耐藥中的研究
2011-09-11 01:24:24祝冰晶周向東
中國肺癌雜志 2011年8期
祝冰晶 周向東
肺癌是當前世界對人類健康和生命威脅最嚴重的腫瘤之一,其發病率和死亡率已躍居各類惡性腫瘤的前列。其致死的主要原因是轉移和耐藥性的出現,這是臨床上治療肺癌的難點,也是肺癌研究的熱點。已有文獻[1]報道磷脂酰肌醇-3-激酶/絲蘇氨酸蛋白激酶(phosphatidylinositol 3-kinase/serine-threonine kinase, PI3K/AKT)信號通路的異常與腫瘤生長、維持和化療耐藥等方面有關。研究[2]表明PI3K/AKT信號通路的失調對腫瘤形成具有重要的作用,多個跨膜受體和配體結合所產生的細胞增殖信號可激活PI3K/AKT信號通路,這與腫瘤細胞的增殖和存活狀態有密切關聯。活化的 PI3K/AKT信號通路在廣泛的人類腫瘤譜中失衡,目前有研究發現肺癌轉移和耐藥都與PI3K/AKT信號轉導通路關系密切,其主要是由于PIK-3CA基因編碼的PI3K擴增和/或其他多種因素導致的AKT過度活化[3],或者是該通路某些調控成分,如類脂磷酸酶(phosphatase and tensin homolog deleted on chromosome ten,PTEN)的突變所導致的功能缺失[4]。因此,對PI3K/AKT信號通路的深入了解有助于找到肺癌治療的潛在靶點。
1 PI3K/AKT信號通路的組成
磷脂酰肌醇3-激酶(phosphoinositide 3-kinase, PI3K)家族成員屬于原癌基因,是肌醇與磷脂酰肌醇(phosphatidyl-inositol, PI)的重要激酶,是細胞內重要的信號轉導分子,參與調節細胞增殖、凋亡與分化等過程,使PI環上的3’羥基磷酸化[5,6]。PI3K可分為3種類型,即I型、II型和III型。I型PI3K以PI、磷脂酰肌醇-4-磷酸(phosphatidylinositol 4-phosphate, PIP)及磷脂酰肌醇-4,5-二磷酸(phosphatidyl-inositol 4,5-biphosphate, PIP2)為底物,使底物的肌醇環第3位發生磷酸化;II型PI3K在羧基末端有C2結構域,主要磷酸化PI和PIP;III型PI3K由催化亞基Vps34和調節亞基p150構成,以PI為底物,主要參與調控細胞生長與存活。目前研究最廣泛的是能被細胞表面受體活化的I型PI3K。I型PI3K又分為IA和IB兩個亞型,分別從酪氨酸蛋白激酶偶聯受體和G蛋白偶聯受體傳遞信號[7]。PI3K可被受體酪氨酸激酶和非受體酪氨酸激酶活化,在細胞膜上生成PIP3。PIP3與細胞內磷酸肌醇依賴性蛋白激酶1(3-phosphoinositide-dependent protein kinase-1, PDK1)和信號蛋白分子AKT(又名蛋白激酶B)結合,從而活化AKT。
AKT是一種絲氨酸/蘇氨酸激酶,約由480個氨基酸殘基組成,與蛋白激酶A(protein kinase A, PKA)和蛋白激酶C(protein kinase C, PKC)高度同源,是PI3K主要下游效應分子之一,可通過直接磷酸化多種轉錄因子如NF-κB和哺乳類動物雷帕霉素(the mammalian target of rapamycin, mTOR)等參與調節多種生命活動過程[8]。AKT分子氨基酸組成由N端到C端依次為PH結構域、中心催化結構域和短羧基端調節結構域[9-11]。PH結構域約含100個氨基酸殘基,介導AKT活化后的膜轉位過程;催化結構域含ATP結合位點,結構域內部Thr308的磷酸化為AKT活化所必需;C端調節域富含脯氨酸,并有AKT活化所必需的另一個磷酸化位點Ser473。目前發現AKT家族成員有3種亞型,包括AKT1/PKBα、AKT2/PKBβ和AKT3/PKBγ,分別由3個不同基因編碼,但蛋白質高級結構基本相同,在各種組織中均廣泛表達。
2 PI3K/AKT信號通路的調控
PI3K/AKT信號轉導通路受多因子調節,參與調節的分子主要是負反饋分子,包括PTEN、CTMP(carboxylterminal modulator protein)、PHLPP(PH domain leucinerich repeat protein phosphatase)和SHP2(Src homology phosphotyrosyl phosphatase 2)等抑癌蛋白。PTEN可拮抗PI3K/AKT信號通路,催化PIP3去磷酸化生成PIP2,從而拮抗AKT的活性[12,13]。在多種腫瘤組織中均能檢測到PTEN的缺失或突變, PI3K信號通路則相應顯現高活性。PHLPP能特異的使AKT在Ser473位點去磷酸化,從而負調控AKT的活性[14]。
許多對PI3K正性調節的研究集中于其上游的活化過程,包括生長因子在內的各種刺激能依賴受體/非受體酪氨酸激酶來激活PI3K[15-17]。胞漿PIP3濃度升高,作為第二信使與AKT的PH結構域結合,使AKT從細胞質轉位到細胞膜以及構象改變發生磷酸化而被激活。活化的AKT再進一步激活其下游分子,如mTOR、Bcl-2家族、轉錄因子E2F-1、糖原合成酶激酶-3(glycogen synthase kinase 3,GSK3)和S6蛋白激酶等,從而對細胞增殖、凋亡和血管生成等進行調節(圖1)。但PI3K通路并非AKT激活的惟一途徑,有研究[18]表明EGF能促進細胞內Ca2+釋放,增加胞漿內Ca2+水平,鈣調蛋白是通過直接與AKT結合來調控其活性的。
3 PI3K/AKT信號通路與肺癌的轉移
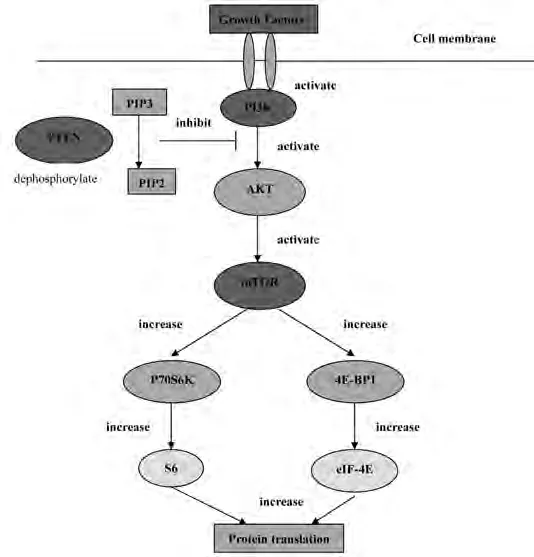
圖 1 PI3K/AKT/mTOR軸[19]Fig 1 PI3K/AKT/mTOR axis[19]
惡性腫瘤的轉移機制涉及腫瘤細胞遺傳物質、表面結構、侵襲力、黏附能力、血管新生、淋巴管新生等多種因素。國內外研究證實PI3K/AKT信號通路與肺癌的轉移密切相關。PI3K/AKT信號通路促使肺癌發生轉移的機制主要有3種:①影響腫瘤黏附能力。上皮細胞-間充質細胞轉換(epithelial-mesenchymal transition, EMT)可使上皮細胞獲得纖維母細胞樣特性,降低細胞間黏附力,增強運動能力。在細胞外信號分子與細胞膜表面的特異性受體結合后,通過細胞內不同信號轉導途徑活化,獲得不同的轉錄因子,使上皮細胞表型發生不同程度的轉化。有研究[20,21]報道PI3K/AKT信號通路參與誘導鱗癌細胞EMT的發生:活化的PI3K產生第二信使PIP3活化下游的AKT,進而通過磷酸化作用激活或抑制下游靶蛋白,調節細胞的生存、增殖分化及細胞骨架的構成等,誘導EMT的發生,PI3K/AKT信號通路的活化增加了腫瘤細胞的侵襲性和轉移性。Steelman等[22]認為,PI3K/AKT的持續活化及高表達與非小細胞肺癌(non-small cell lung cancer, NSCLC)發生EMT關系密切。②對腫瘤新生血管的影響。腫瘤的轉移擴散依賴于新生血管形成。目前已發現多條信號通路參與調控腫瘤血管生成,PI3K/AKT是其中極重要的一條[23]。PI3K通過與E-Cadherin、β-Catenin、和血管內皮生長因子受體-2(vascular endothelial growth factor receptor 2, VEGFR-2)形成復合物由PI3K/AKT通路的活化參與血管內皮生長因子(vascular endothelial growth factor, VEGF)介導的內皮信號傳遞[24,25]。PI3K/AKT信號通路還能促進腫瘤壞死因子(tumor necrosis factor, TNF)誘導的內皮細胞遷移,調節腫瘤新生血管生成[26]。基質金屬蛋白酶(matrix metalloproteinases, MMPs)和環氧化酶-2(cyclooxygenase-2, COX-2)也能影響腫瘤新生血管的形成。在腫瘤的侵襲轉移中,血小板源性生長因子(plateletderived growth factor, PDGF)經過由PI3K介導的信號通路誘導MMPs表達[27],上調抗凋亡蛋白Bcl-2和激活PI3K/AKT信號通路是COX-2刺激內皮細胞血管生成的主要機制[28,29]。由此可見,PI3K/AKT信號通路參與了多種因子介導的腫瘤新生血管的生成。③通過PI3K/AKT/mTOR/p70s6k途徑,促進肌動蛋白的細絲重構,促進腫瘤細胞運動轉移[30]。
4 PI3K/AKT信號通路與肺癌耐藥
NSCLC約占肺癌總發病率的85%,放療、化療是其主要治療手段,但腫瘤細胞對化療藥物耐受性的產生是導致肺癌化療失敗,患者生存率低的主要原因之一。有研究[31]表明目前認為以鉑類為主的聯合化療在NSCLC的治療中有效率為50%左右,被推薦為治療NSCLC的標準方案,不僅可減輕癥狀、提高生存質量,還可延長生存期。順鉑(cis-dichlorodiamine platinum, CDDP)是肺癌化療中有效且廣泛應用的一線藥物,但是由于肺癌細胞耐藥性問題的存在,其療效不盡人意。CDDP等化療藥物不僅能夠直接殺傷腫瘤細胞,還可以通過誘導腫瘤細胞發生凋亡而發揮作用[32]。抑制凋亡是腫瘤細胞產生耐藥性的共同途徑,細胞進入凋亡程序的能力可直接影響化療藥物發揮作用的效應。腫瘤細胞通過抑制進入凋亡程序表現出對化療藥物的耐受性,是腫瘤細胞的自我保護功能之一。選擇性誘導腫瘤細胞發生凋亡已經成為腫瘤治療的一條有效途徑而倍受關注,并可能明顯改善患者的預后[33]。PI3K/AKT信號轉導通路能夠調節細胞生存、增殖和分化等多種功能,是最主要的抑制細胞凋亡的信號途徑[34,35]。Liu等[36]研究證實,PI3K/AKT信號通路與肺癌細胞對CDDP的耐藥密切相關,通過抑制AKT1的表達可有效逆轉肺癌細胞對CDDP的耐藥,采用PI3K的抑制劑LY294002能明顯抑制AKT1的表達并增強肺癌耐藥細胞對CDDP的敏感性。
PI3K/AKT信號轉導通路抑制細胞凋亡的機制為:①通過磷酸化forkhead家族轉錄因子阻止其發揮調節相關基因的轉錄功能[37];②活化轉錄因子NF-κB,NF-κB進入細胞核后,促進細胞增殖,抑制細胞凋亡,從而誘導抗凋亡基因Bcl-2、Bcl-XL的表達;③活化的AKT直接磷酸化cAMP應答元件結合蛋白(cAMP response element binding protein, CREB)的Ser133位點,誘導Bcl-2、Bcl-XL等相關基因的表達;④PI3K依賴的AKT激活,使促細胞凋亡基因Bad與Bcl-2或Bcl-xL解聚,Bad再與抗凋亡結合蛋白14-3-3相結合,而游離的Bcl-2發揮抗凋亡作用[38];⑤抑制天冬氨酸特異性半胱氨酸蛋白酶家族成員的活化,活化的AKT可以直接催化磷酸化caspase-9的Ser196和caspase-3,使其失活從而抑制caspase導致的細胞凋亡;⑥抑制腫瘤細胞膜上TRAIL受體的活性,從而抑制由凋亡受體家族介導的細胞凋亡[39];⑦通過調節細胞周期影響細胞增殖,mTOR是AKT下游的一個重要作用靶點,能夠被AKT磷酸化激活,調控相關mRNA的轉錄,進而調節蛋白質的合成,影響細胞的增殖,AKT1誘導肺癌細胞對CDDP的耐藥是通過mTOR-P70核糖體S6激酶(P70S6K1)信號途徑進行[36]。
腫瘤分子的靶向治療研究發現信號轉導通路與腫瘤進展以及放、化療抵抗密切相關。PI3K/AKT信號通路在肺癌細胞耐藥中發揮了非常重要的作用[36]。研究[40,41]表明PI3K/AKT的抑制劑在體外可抑制腫瘤細胞的生長,誘導腫瘤細胞進入凋亡程序,對于AKT高表達的NSCLC,使用PI3K/AKT信號通路的抑制劑能夠增強化療誘導的癌細胞凋亡,減少化療抵抗,抑制PI3K/AKT信號通路能夠有效地提高藥物誘導的肺癌細胞凋亡。
5 PI3K/AKT信號通路抑制劑的研究
PI3K/AKT信號通路在肺癌發生、發展至關重要的眾多細胞生物學過程都發揮了重要作用,抑制該信號通路成為了肺癌預防和靶向治療的熱點。PI3K/AKT信號通路的各個激酶,均有多種抑制劑處于臨床前、臨床研究及應用階段。目前已經發現了該信號通路中多種激酶的小分子抑制劑(small molecule inhibitors, SMIs),其中Wortmannin和LY294002是兩種廣泛應用的PI3K/AKT信號通路的抑制劑。Wortmannin可與PI3K相對分子質量1.1×105催化亞基結合,特異性抑制PI3K,從而抑制PI3K/AKT信號通路。PX-866和PWT-458是近年新發現的Wortmannin的衍生物,可高效抑制PI3K。在肺癌模型中PX-866與順鉑或放療聯合使用能增強抗腫瘤作用[42]。此外,PWT-458還能增強紫杉醇抗腫瘤的療效[43]。LY294002可完全特異性的抑制PI3K的活性,能和PI3K競爭性結合ATP位點,抑制AKT的活性。LY294002與常規放化療聯合應用具有協同作用,并可以減少不良反應,對標準化方案產生耐藥的腫瘤可考慮聯合應用PI3K抑制劑[44]。吳等[45]發現LY294002可抑制A549細胞增殖,且隨LY294002濃度的增大及作用時間的延長,生長抑制率也隨之增大;同時LY294002可抑制裸鼠移植瘤的生長,使移植瘤的磷酸化AKT蛋白表達下降,提示LY294002可通過抑制PI3K活性,進而調控AKT表達及活化,從而抑制肺癌細胞的生長。研究[46-48]表明Wortmannin和LY294002均能通過抑制AKT磷酸化進而發揮抗腫瘤的效應。
PI3K抑制劑在NSCLC的研究中證明在體內單獨使用及與化療藥物聯合應用均能增加腫瘤細胞的凋亡[49]。磷酸化的AKT通過將信號傳遞給眾多下游分子參與腫瘤發生發展,是目前腫瘤靶向研究的一個熱點。抑制AKT的活性能抑制下游靶基因轉錄,從而抑制下游信號傳遞。目前研究的AKT抑制劑主要有兩種形式:脂類抑制劑和通過高通量篩選化合物庫所得的SMIs。脂類抑制劑有perifosine、磷脂酰肌醇烷脂和PX-316。對于perifosine的研究最為透徹,能抑制AKT的膜轉位,并能通過降低AKT的活性抑制多種腫瘤細胞生長[50]。SMIs主要有API-2、API-59CJ-Ome和吲哚-嘧啶A-443654等。
6 結語
細胞生存信號的轉導對肺癌的發生發展非常重要,而PI3K/AKT信號通路被認為是癌細胞存活的首要通路。目前對PI3K/AKT信號通路的作用及其具體調節機制認識尚不完全,許多問題還有待進一步探討。隨著對PI3K/AKT信號轉導通路及其在肺癌中重要作用研究的逐漸深入,針對該信號通路開發新的腫瘤治療藥物對肺癌的治療具有十分重要的意義。
近年以PI3K/AKT信號通路分子作為靶點的抗腫瘤藥物已得到很好的開發,這些AKT特異性抗腫瘤藥物可拮抗AKT抗凋亡作用,破壞癌細胞的耐藥性,誘導癌細胞發生凋亡[51]。此外,還能有效提高化療藥物順鉑、紫杉醇對體外肺癌細胞抑制作用的敏感性,抑制PI3K/AKT信號通路可明顯提高肺癌的化療效果,減少化療藥物的劑量,提高患者對治療的依從性。但是特異性抑制劑因其毒性較強使臨床應用具有一定限制性,此外,在基礎研究及動物模型取得的抗癌效果對于肺癌病例是否具有足夠的效力而改變癌癥的進展及PI3K/AKT信號通路的下游底物并未完全了解,還有待進一步研究。PI3K/AKT信號通路的任何一個環節的異常都與肺癌的發生發展密切相關。因此,全面、系統地深入研究PI3K/AKT信號轉導通路分子機制對以信號通路作為靶點的肺癌防治工作具有非常重要的意義。
猜你喜歡
保健醫苑(2023年2期)2023-03-15 09:03:04
中國臨床醫學影像雜志(2022年2期)2022-05-25 13:24:34
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
中國生殖健康(2019年3期)2019-02-01 06:12:26
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
癌變·畸變·突變(2016年3期)2016-02-27 06:15:34
海軍航空大學學報(2015年3期)2015-11-11 17:20:00
醫學研究雜志(2015年12期)2015-06-10 06:57:46
鄭州大學學報(醫學版)(2015年1期)2015-02-27 14:50:26
