環(huán)己烷氣相氧化脫氫催化劑的研究進展
2011-07-25 05:44:04余國賢李忠銘
化學與生物工程 2011年3期
關(guān)鍵詞:催化劑
晉 梅,余國賢,李忠銘
(江漢大學化學與環(huán)境工程學院,湖北 武漢 430056)
1 引言
環(huán)己烯是一種重要的有機化工原料,廣泛用于醫(yī)藥、食品、農(nóng)用化學品、飼料、聚酯等精細化工產(chǎn)品的生產(chǎn)。自Sato等[1]從環(huán)己烯直接氧化合成己二酸后,環(huán)己烯就被認為是合成環(huán)己酮、環(huán)己醇和己二酸的最佳原料[2,3]。環(huán)己烯可通過兩種方法獲得:一是目前工業(yè)上采用的苯選擇加氫反應(yīng),然而其轉(zhuǎn)化率低、選擇性不高、副產(chǎn)物環(huán)己烷較多且難以利用[4];二是環(huán)己烷脫氫反應(yīng),但脫氫生成苯是熱力學穩(wěn)定體系[5]。因此,環(huán)己烷氣相氧化脫氫制環(huán)己烯將成為生產(chǎn)己二酸的新途徑,也是充分利用苯部分加氫副產(chǎn)物環(huán)己烷以形成環(huán)己烷-苯-環(huán)己烯循環(huán)利用并最大化生產(chǎn)環(huán)己烯的綠色工藝路線。
環(huán)己烷氣相氧化脫氫反應(yīng)由一系列平行串聯(lián)反應(yīng)組成且在較高反應(yīng)溫度下進行,目的產(chǎn)物環(huán)己烯為反應(yīng)的中間產(chǎn)物,存在著環(huán)己烷轉(zhuǎn)化率和環(huán)己烯選擇性之間的矛盾。反應(yīng)的關(guān)鍵是解決反應(yīng)過程中轉(zhuǎn)化率和選擇性之間的協(xié)調(diào)問題以及催化劑在高溫下的穩(wěn)定性問題。為了解決這一問題,一個切實可行的方法是采用可活化環(huán)己烷分子中C-H鍵同時避免生成C-O鍵的催化劑。近年來,國內(nèi)外針對環(huán)己烷氧化脫氫反應(yīng)催化劑體系的研究主要集中在陽離子沸石催化劑、復合金屬氧化物催化劑以及貴金屬絲網(wǎng)催化劑等體系,致力于在較緩和的反應(yīng)條件、較易的催化劑制備方法下獲得較高的環(huán)己烯產(chǎn)率和較少的COx排放。作者在此對上述三類催化劑體系所遵循的反應(yīng)機理、催化性能和反應(yīng)條件進行詳細的闡述,同時對金屬氧化物催化劑體系中的釩基、鉬基以及鎳基催化劑的反應(yīng)活性中心進行進一步探討,為今后環(huán)己烷氧化脫氫反應(yīng)催化劑的研究提供參考。
2 環(huán)己烷氣相氧化脫氫反應(yīng)機理
對不同催化劑體系的氧化脫氫反應(yīng)機理進行探討,尋找反應(yīng)速控步驟,對提高環(huán)己烯的選擇性和收率具有十分重要的意義。
2.1 自由基反應(yīng)機理
Alimardanov等[6]采用沸石載體金屬氧化物為催化劑,研究環(huán)己烷氧化脫氫反應(yīng)機理,如式(1)~(9)所示。首先,環(huán)己烷在催化劑表面生成環(huán)己基自由基,繼而脫氫生成環(huán)己烯,同時伴隨生成小分子烴及COx的副反應(yīng)。
(1)
(2)
C6H12+0.5O2→C6H10+H2O
(3)
C6H10+O2→C6H6+2H2O
(4)
C6H10+1.5O2→C5H8+CO2+H2O
(5)
C6H10+2.5O2→C4H8+2CO2+H2O
(6)
C6H10+4O2→C3H6+3CO2+2H2O
(7)
C6H10+5.5O2→C2H4+4CO2+3H2O
(8)
C6H10→C5H7CH3
(9)
2.2 氧化還原反應(yīng)機理
遵循Mars-van Krevelen反應(yīng)機理的氧化物有V2O5、MoO3、Bi2O3以及Bi2O3-MoO3等,具有反應(yīng)溫度較高、環(huán)己烯選擇性高的特點。反應(yīng)機理為:(1)氣相環(huán)己烷分子和氧氣分別在催化劑表面進行吸附;(2)吸附于催化劑表面的氣相烴分子與高價態(tài)金屬氧化物催化劑表面上的晶格氧相互作用,烴分子被氧化為環(huán)己基自由基;晶格氧參與反應(yīng)后,催化劑上的金屬氧化物被還原為較低價態(tài);(3)氣相氧將低價態(tài)的金屬氧化物氧化到初始高價態(tài),補充晶格氧,完成催化劑上金屬陽離子的氧化-還原循環(huán)。反應(yīng)速控步驟為吸附的環(huán)己烷分子中-H的提取。
2.3 均相氣相反應(yīng)和非均相氣相反應(yīng)共存的反應(yīng)機理
O′Connor等[11,12]在貴金屬的單一金屬絲網(wǎng)反應(yīng)器中對環(huán)己烷氧化脫氫反應(yīng)進行研究,提出反應(yīng)機理,如圖1所示。
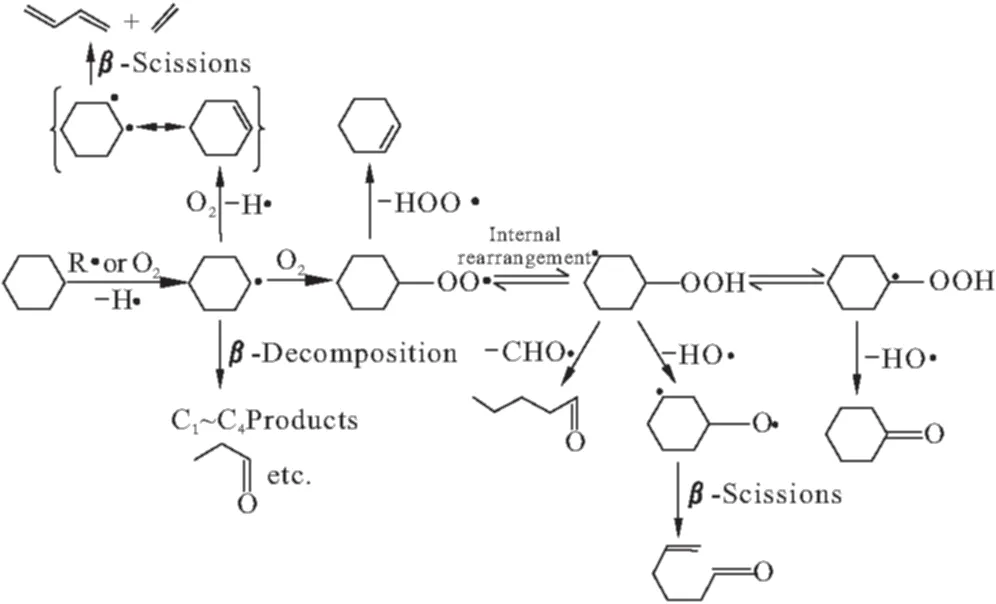
圖1 環(huán)己烷部分氧化的反應(yīng)路徑
環(huán)己烷分子首先進行非均相氣相反應(yīng)生成環(huán)己基自由基,而后進一步脫氫生成環(huán)己烯。整個反應(yīng)體系過程在高溫及毫秒級接觸時間下進行,其中還伴隨有均相氣相反應(yīng),產(chǎn)物中有大量氧化副產(chǎn)物,環(huán)己烯分離極其困難。
3 環(huán)己烷氧化脫氫制備環(huán)己烯的催化劑性能
不同催化劑體系在環(huán)己烷氧化脫氫反應(yīng)中遵循不同的反應(yīng)機理,因而在環(huán)己烷氧化脫氫的反應(yīng)體系中采用的操作條件和催化性能有較大差異,如表1所示。
從表1可知:(1)環(huán)己烷氧化脫氫反應(yīng)的氧化劑是低濃度的氧氣或空氣,不僅具有適宜的氧化性且價廉易得;(2)環(huán)己烷氧化脫氫反應(yīng)的溫度主要集中在400~600℃;(3)貴金屬絲網(wǎng)催化劑的反應(yīng)的條件最為苛刻,反應(yīng)溫度高達900°C、接觸時間<5 ms;(4)采用復合金屬氧化物作為催化劑進行環(huán)己烷氧化脫氫反應(yīng)的條件相對較為溫和且催化性能相對較好,針對該體系的研究也最多。
4 金屬氧化物催化劑催化活性組分
環(huán)己烷氧化脫氫反應(yīng)中,研究最多的金屬氧化物催化劑體系主要集中在釩基、鉬基和鎳基氧化物。
4.1 釩基催化劑
雖然V5+具有很好的活化烴分子上C-H鍵的能力[29,30],但其結(jié)構(gòu)中V=O有利于C-O鍵的生成,產(chǎn)物中COx量較多。為了提高目的產(chǎn)物環(huán)己烯的產(chǎn)率和選擇性,通常向V2O5中加入另一種金屬陽離子以形成金屬釩酸鹽。釩酸鹽結(jié)構(gòu)中的V-O-Me鍵會改變V-O活性中心的電子云密度分布、催化劑表面的酸堿性和活性物種的氧化還原性速率。比較典型的金屬陽離子有兩種:一種電負性小于釩,如Mg;另一種電負性大于釩,如P。
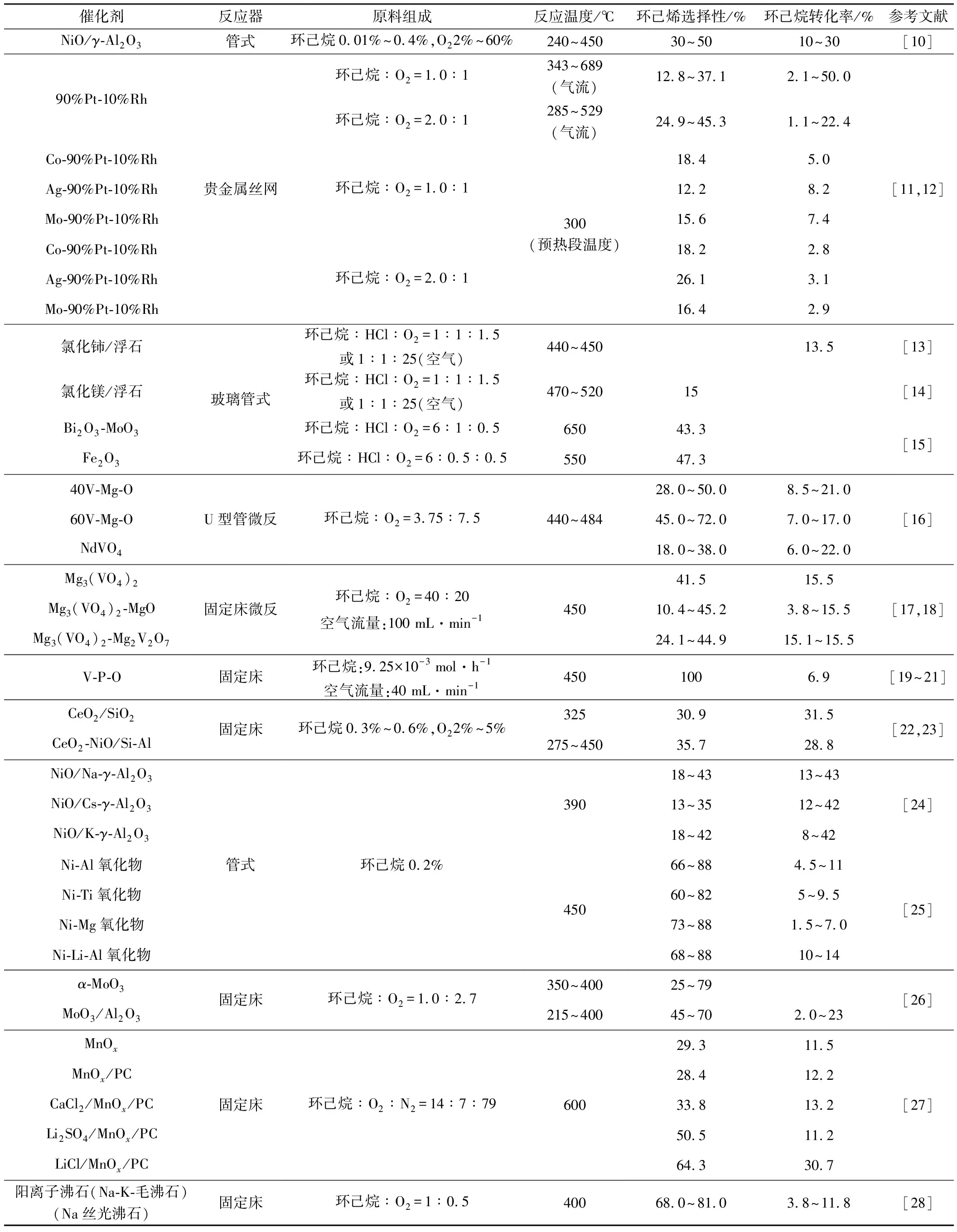
表1 環(huán)己烷氧化脫氫反應(yīng)催化劑的性能
根據(jù)V/Mg原子比的不同,V-Mg-O催化劑主要以Mg3(VO4)2、Mg2V2O7與MgV2O6三種穩(wěn)定形式存在。在環(huán)己烷氧化脫氫反應(yīng)中,Kung等[16]采用共沉淀法制備40(60)V-Mg-O催化劑[Mg3(VO4)2+MgO+V2O5],雖取得較好催化效果,但未對催化活性相進行研究。Grasselli等提出Mg3(VO4)2晶相具有“孤立活性位”、適量的晶格氧含量、催化劑表面弱堿性以及較低的金屬離子可還原性,可作為環(huán)己烷氧化脫氫反應(yīng)的催化活性相[31~33]。方智敏等[34,35]對V-Mg-O進行原位Raman光譜研究表明:V4+?V5+氧化還原對易于形成氧缺位而有利于高溫吸附并活化氧分子,轉(zhuǎn)化為晶格氧以參與環(huán)己烷氧化脫氫反應(yīng),且Mg3(VO4)2、Mg2V2O7和MgV2O6的表面活性位為V4+-O-Mg-O-V5+、V4+-O-V5+和V5+-O。其中V4+-O四面體配位結(jié)構(gòu)是氧化脫氫反應(yīng)的活性位,有利于環(huán)己烯選擇性的提高;V5+-O八面體配位結(jié)構(gòu)是加氧氧化反應(yīng)的活性位[36]。
在Carrazán等[37]和Gao等[38]研究的基礎(chǔ)上,Jin等[17]對催化活性相與其它晶相間的協(xié)同效應(yīng)進行了研究。他們認為,在Mg3(VO4)2活性相中加入適量的MgO時,固溶的MgO不僅占據(jù)環(huán)己烷氧化脫氫反應(yīng)的催化活性位且增強了催化劑的表面堿性;當加入Mg2V2O7時,晶相間由于溢流氧產(chǎn)生的遙控機理作用而促進溢流氧從供體遷移到受體上并與受體作用以修飾催化活性位。類似地,Pantazidis等[39]認為過量的MgO不僅可調(diào)節(jié)催化劑的氧化還原性能,同時可改變催化劑表面酸堿性,并提出適度增強催化劑的堿性有利于V4+的生成、不利于表面碳酸鹽的生成。
P的電負性大于釩,因此催化劑表面親核性變差而導致催化劑表面呈現(xiàn)弱酸性。與V-Mg-O催化劑相同,V-P-O晶相結(jié)構(gòu)中由于PO4基團與VO6基團共享的氧所處結(jié)構(gòu)不同[40],通常以三個穩(wěn)定的晶相結(jié)構(gòu)形式存在:αI-VOPO4、αⅡ-VOPO4和β-VOPO4。Herrmann等[41]研究V-P-O催化劑導電性發(fā)現(xiàn),V-P-O屬于p型半導體材料,主要電荷載體為正電荷空穴P+,且若要達到最佳催化效果必須有V5+存在。活性位的氧化-還原通過(VO)3+?(VO)2++P+和P++O2-?O-來完成,參與反應(yīng)的氧物種為晶格氧物種,這一結(jié)論在Abon等[42]用18O2同位素進行的氧化脫氫反應(yīng)實驗中得到了驗證。朱宇君等[19~21]僅采用αⅠ-VOPO4為環(huán)己烷氧化脫氫反應(yīng)催化劑時,產(chǎn)物中并沒有出現(xiàn)環(huán)己烯,究其原因主要為:一方面αⅠ-VOPO4呈弱酸性,不利于中間產(chǎn)物環(huán)己烯的脫附;另一方面,V5+的存在可使得催化劑達到最佳催化效果,但又利于深度氧化反應(yīng)的進行。為了提高環(huán)己烯的選擇性,他們采用醋酸共進料的方法,通過醋酸分子的-OH與αⅠ-VOPO4晶相中V=O形成氫鍵吸附在催化劑表面,形成孤立的活性位點。
4.2 鉬基催化劑
MoO3為八面體結(jié)構(gòu)的酸性、非嚴格化學計量氧化物,其內(nèi)部結(jié)構(gòu)存在大量氧空穴,結(jié)構(gòu)缺陷和氣相氧之間通過Mo5+?Mo6+氧化還原偶保持動態(tài)平衡。其中Mo6+八面體結(jié)構(gòu)利于深度氧化反應(yīng),Mo5+不飽和配位結(jié)構(gòu)則有利于烯烴選擇性的提高。Cadus等[43]對Mg-Mo-O催化劑進行EPR表征,發(fā)現(xiàn)較高Mo5+含量的催化劑在氧化脫氫反應(yīng)中具有好的催化性能且MoO3稍過量利于Mo5+物種的形成。同時,Allison等[44]提出了含有相鄰表面雙氧位的催化活性位的反應(yīng)活化能低于單一鉬雙氧位的熱力學和量子力學結(jié)果,利于烯烴選擇性的提高。Alyea等[26]采用金屬氧化物氣相合成方法制備出含雙重二氧鉬活性位的α-MoO3及MoO3/Al2O3催化劑用于環(huán)己烷氧化脫氫反應(yīng),主要的催化活性位為Mo5+物種。
4.3 鎳基催化劑
5 結(jié)語
環(huán)己烷氧化脫氫反應(yīng)催化劑研究雖取得了一定的進展,但仍然存在很多亟待解決的問題,如催化劑活性與選擇性的協(xié)調(diào)、氧物種類型的選擇、工業(yè)應(yīng)用等。因此,開發(fā)新型催化劑體系、新的環(huán)己烷氧化脫氫反應(yīng)途徑勢在必行。工藝應(yīng)用研究、反應(yīng)器的設(shè)計以及反應(yīng)器中傳熱傳質(zhì)問題的研究還需進一步加強。
總之,環(huán)己烷氧化脫氫制取環(huán)己烯不僅為環(huán)己烯的合成開辟了一條新的途徑,且為高效利用來源豐富、價格便宜的環(huán)己烷提供了新的思路,具有廣闊的工業(yè)應(yīng)用前景。
[1]Sato K,Aoki M,Noyori R.A "green" route to adipic acid:Direct oxidation of cyclohexenes with 30 percent hydrogen peroxide [J].Science,1998,281(11):1646-1648.
[2]Schuchardt U,Cardoso D,Sercheli R,et a1.Cyclohexane oxidation continues to be a challenge[J].Appl Catal A:General,2001,211(1):1-17.
[3]宮紅,姜恒,呂振波.己二酸綠色合成新途徑[J].高等學校化學學報,2000,21(7):1121-1123.
[4]Liu S C,Liu Z Y,Wang Z,et al.Characterization and study on performance of the Ru-La-B/ZrO2amorphous alloy catalysts for benzene selective hydrogenation to cyclohexene under pilot conditions[J].J Chem Eng,2008,139(1):157-164.
[5]Riad M,Mikhail S.Dehydrogenation of cyclohexane over molybdenum/mixed oxide catalysts[J].Catal Comm,2008,9(6):1398-1403.
[6]Alimardanov K M.Kineties and mechanism of cyelohexane dehydrogenation to cyclohexene in the presence of molecular oxygen[J].J Petroleum Chemistry:USSR (English Translation of Neftekhimiya),1991,31(1):22-29.
[7]Bielan′ski A,Haber J.Oxygen in catalysis on transition metal oxides[J].Catal Rev Sci Eng,1979,19(1):1-41.
[8]辛勤.固體催化劑研究方法[M].北京:科學出版社,2004:245-250.
[9]Sokolovskii V,Arena F,Giordano N,et al.Role of acid-base properties of SiO2-based catalysts in the selective oxidation of propane[J].J Catal,1997,167(1):296-299.
[10]Patcas F,Pateas F C.Reaction pathways and kinetics of the gas-phase oxidation of cyclohexane on NiO/γ-Al2O3catalyst[J].Catal Today,2006,117(1):253-258.
[11]O′Connor R P,Klein E J,Henning D,et al.Tuning millisecond chemical reactors for the catalytic partial oxidation of cyclohexane[J].Appl Catal A:General,2003,238(1):29-40.
[12]O′Connor R P,Schmidt L D.Catalytic partial oxidation of cyclohexane in a single-gauze reactor[J].J Catal,2000,191(1):245-256.
[13]Tallman R C,Ark E D.Conversion of cyclohexane to cyclohexene[P].USP 3 108 144,1963-10-22.
[14]Boonton N J,Morristown N J.Producing cyclohexene or alkycyclohexene[P].USP 4 356 337,1982-10-26.
[15]趙紅坤,雒廷亮,楊杰,等.環(huán)己烷氧化脫氫制備環(huán)己烯的研究現(xiàn)狀及展望[J].浙江化工,2002,33(4):51-54.
[16]Kung H H,Kung M C.Oxidative dehydrogenation of alkanes over vanadium-magnesium oxides[J].Appl Catal A:General,1997,157(1-2):105-116.
[17]Jin M,Cheng Z M.Oxidative dehydrogenation of cyclohexane to cyclohexene over Mg-V-O catalysts[J].Catal Lett,2009,131(3-4):266-278.
[18]Jin M,Cheng Z M,Gao Y L,et al.Oxidative dehydrogenation of cyclohexane with Mg3(VO4)2synthesized by the citrate process[J].Mater Lett,2009,63(23):2055-2058.
[19]朱宇君,李靜,楊向光,等.醋酸對環(huán)己烷氣相氧化脫氫產(chǎn)物選擇性影響的研究[J].高等學校化學學報,2006,27(6):1118-1120.
[20]Zhu Y J,Li J,Yang X G,et al.A new route to control product selectivity in the oxidative dehydrogenation of cyclohexane and cyclohexene[J].Chem Lett,2004,33(7):822-823.
[21]Zhu Y J,Li J,Xiao F X,et al.Effect of different VOPO4phase catalysts on oxidative dehydrogenation of cyclohexane to cyclohexene in acetic acid[J].J Mole Catal A:Chemical,2006,246(1):185-189.
[22]Kubo Toshihiko.Oxidative dehydrogenation of cyclohexane to cyclohexene over zeolite catalyst[J].Nippon Kagaku Kaishi,1973,12:2257-2263.
[23]Hayakawa Takshi.Oxidative dehydrogenation of cyclohexane with Ce catalysts supported on Si[J].J Sekiyu Gakkaishi,1987,30:161-165.
[24]Patcas F,Honicke D.Effect of alkali doping on catalytic properities of alumina-supported nickel oxide in the selective oxidehydrogenation of cyclohexane[J].Catal Comm,2005,6(1):23-27.
[25]Patcas F,Krysmann W,Honicke D,et al.Preparation of structured egg-shell catalysts for selective oxidations by the ANOF technique[J].Catal Today,2001,69(2):379-383.
[26]Alyea E C,Keane1 M A.The oxidative dehydrogenation of cyclohexane and cyclohexene over unsupported and supported molybdena catalysts prepared by metal oxide vapor deposition[J].J Catal,1996,164(1):28-35.
[27]Zhu H,Ge Q J,Li W Z,et al.Study of Mn-based catalysts for oxidative dehydrogenation of cyclohexane to cyclohexene[J].Catal Lett,2005,105(1):29-34.
[28]Dietz I A G,Carlsson A F,Schmidt L D.Partial oxidation of C5and C6alkanes over monolith catalysts at short contact times[J].J Catal,1996,167(2):459-473.
[29]Hutchings G J,Desmartin-Chomel A,Olier J C,et al.Role of the product in the transformation of a catalyst to its active state[J].Nature,1994,368(1):41-42.
[30]Kung H H,Birkeland K,Bethke G K,et al.The kinetic significance of V5+inn-butane oxidation catalyzed by vanadium phosphates[J].Science,1997,275(1):191-193.
[31]Charr M A,Patel D,Kung M C,et al.Selective oxidative dehydrogenation of butane over V-Mg-O catalysts[J].J Catal,1987,105(2):483-498.
[32]Grasselli R K.Fundamental principles of selective heterogeneous oxidation catalysis[J].Topics in Catal,2002,21(1):79-88.
[33]Patel D,Kung M C,Kung H H.Proceeding,9th international congress on catalysis[C].Calgary:Chem Institute of Canada,Ottawa,1988,4:1553-1555.
[34]方智敏,翁維正,萬惠霖.VMgO催化劑上丙烷氧化脫氫反應(yīng)的原位Raman譜學研究[J].分子催化,1998,12(3):207-213.
[35]方智敏,翁維正,萬惠霖.丙烷氧化脫氫VMgO催化劑活性位的研究[J].廈門大學學報(自然科學版),1998,37(4):525-531.
[36]張偉德,沙開清,李基濤,等.丙烷氧化脫氫催化劑V2O5/MPO4(M=A1,Zr,Ca)的研究[J].高等學校化學學報,1999,20(4):608-611.
[37]Carrazán S R G,Peres C,Bernard J P,et al.Catalytic synergy in the oxidative dehydrogenation of propane over MgVO catalysts[J].J Catal,1996,158(2):452-476.
[38]Gao X T,Ruiz P,Xin Q,et al.Effect of coexistence of magnesium vanadate phases in the selective oxidation of propane to propene[J].J Catal,1994,148(1):56-67.
[39]Pantazidis A,Auroux A,Herrmann J M.Role of acid-base redox and structural properties of VMgO catalysts in the oxidative dehydrogenation of propane[J].Catal Today,1996,32(1):81-88.
[40]Volta J C,Olier R,Roullet M,et al.Study byinsitulaser Raman spectroscopy of a VPO catalyst in the course ofn-butane oxidation to maleic anhydride[J].Studies in Surface Science and Catalysis,1993,75:1531-1534.
[41]Herrmann J M,Vernoux P,Béré K E,et al.Insitustudy of redox and of p-type semiconducting properties of vanadyl pyrophosphate and of V-P-O catalysts during the partial oxidation ofn-butane to maleic anhydride[J].J Catal,1997,167(1):106-117.
[42]Abon M,Béré K E,Delichére P.Nature of active oxygen in then-butane selective oxidation over well-defined V-P-O catalysts:An oxygen isotopic labelling study[J].Catal Today,1997,33(1):15-23.
[43]Cadus L E,Gomez M F,Abello M C.Synergy effects in the oxidative dehydrogenation of propane over Mg-MoO4-MoO3catalysts[J].Catal Lett,1997,43(1):229-233.
[44]Allison J N,Goddard W A.Oxidative dehydrogenation of methanol to formaldehyde[J].J Catal,1985,92(1):127-135.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應(yīng)用化工(2014年3期)2014-08-16 13:23:50
