氣相色譜-三重四極桿串聯質譜法測定發酵食品中5種N-亞硝胺類化合物
2025-02-01 00:00:00馬嘉俊傅春輝楊瑞平恬敏傅華靖劉鎮
食品安全導刊 2025年1期
關鍵詞:氣相色譜

摘 要:目的:建立氣相色譜-三重四極桿串聯質譜測定發酵食品中5種N-亞硝胺類化合物的分析方法。方法:樣品經椰殼活性炭固相萃取小柱凈化富集,采用大體積程序升溫進樣,經DB-1701色譜柱分離,采用多反應監測模式進行測定,以質量數和保留時間定性,內標法定量。結果:5種N-亞硝胺類化合物的濃度在25~1 000 μg·L-1線性關系良好,相關系數均大于0.996 0;方法檢出限在0.1~0.8 μg·kg-1,定量限在0.3~
2.8 μg·kg-1;樣品的加標回收率為76.2%~112.1%,相對標準偏差在2.1%~8.1%。結論:該方法準確、可靠,適用于發酵食品醬油、黃酒中5種N-亞硝胺類化合物的檢測。
關鍵詞:N-亞硝胺;發酵食品;氣相色譜-三重四極桿串聯質譜法
Determination of 5 N-Nitrosamine Compounds in Fermented Foods by Gas Chromatography-Triple Quadrupole Tandem Mass Spectrometry
MA Jiajun, FU Chunhui, YANG Rui, PING Tianmin, FU Huajing, LIU Zhen*
(Shaoxing Testing Institute of Food and Drug, Shaoxing 312000, China)
Abstract: Objective: To establish a method for the determination of 5 N-nitrosamines in fermented food by gas chromatography-triple quadrupole tandem mass spectrometry. Method: The samples were purified and concentrated by a small column of solid phase extraction of coconut shell activated carbon, injected with a large volume temperature program, separated by DB-1701 chromatographic column, determined by multiple reaction monitoring mode, qualitative by mass number and retention time, quantitative by internal standard method. Result: The concentration of 5 N-nitrosamines was in the range of 25 μg·L-1 to 1 000 μg·L-1, and the correlation coefficient was greater than 0.996 0. The limits of detection and quantitation were 0.1 μg·kg-1 to 0.8 μg·kg-1 and 0.3 μg·kg-1 to
2.8 μg·kg-1. The recoveries ranged from 76.2% to 112.1%, and the relative standard deviation was 2.1% to 8.1%. Conclusion: The method is accurate and reliable, and is suitable for the determination of 5 N-nitrosamines in fermented food soy sauce and rice wine.
Keywords: N-nitrosamine; fermented food; gas chromatography-tandem triple quadrupole mass spectrometry
在一定條件下,食品中亞硝酸鹽與胺類化合物發生亞硝基化作用生成N-亞硝胺[1]。N-亞硝胺具有強烈的致癌性,可對人體健康造成巨大威脅。因此,對食品與環境中N-亞硝胺污染水平的把控與監測十分必要[2-5]。
食品是人體攝入N-亞硝胺的主要來源,目前食品中N-亞硝胺檢測研究采用的方法包括氣相色譜-熱能分析法(Gas Chromatography-Thermal Energy Analysis,
GC-TEA)、氣相色譜-串聯質譜法(Gas Chromatography-
Tandem Mass Spectrometry,GC-MS/MS)[6]及高效液相色譜-串聯質譜法(High-Performance Liquid Chromatography-Tandem Mass Spectrometry,HPLC-MS/MS)[7-8]等。
熱能分析法對亞硝胺具有高度特異性,但僅局限于亞硝基化合物的分析,且價格昂貴,在推廣應用上存在局限[9]。質譜法利用碎片分析可以定性,色譜-質譜聯用技術結合了色譜高效的分離能力、質譜法精準確定分子量和分子結構的能力,在分析和鑒定復雜多組分有機混合物上應用廣泛。然而,HPLC-MS/MS對低質量數N-亞硝胺存在響應不足的問題。近年來,隨著三重四極桿質譜聯用儀的發展,GC-MS/MS已逐漸成為N-亞硝胺分析的常規檢測手段,采用多反應監測模式掃描,具有選擇性好、定性特異性強、定量準確度高的優點[10-12]。
程序升溫大體積進樣技術(Programmed Temperature
Vaporization,PTV),基于GC-MS技術發展而來,其核心機制在于利用低溫進樣口蒸發樣品中的大部分溶劑,保留低揮發性的微量目標物,再通過程序化升溫使這些物質蒸發,進而進入色譜柱進行分析[13]。傳統的毛細管氣相色譜的柱容量較小,僅能接受微升級(一般在1 μL左右)的進樣量。大體積進樣可將進樣量增加100倍或更多,提高了檢測效率和分析靈敏度[14]。李登昆等[15]將大體積程序升溫進樣應用于飲用水中3種揮發性N-亞硝胺的測定,研究發現大體積程序升溫氣化進樣可通過增加進樣量來提高方法的靈敏度。
目前,關于發酵食品(黃酒、醬油)中N-亞硝胺檢測的研究較少[16]。黃酒和醬油是我國的傳統發酵食品,在發酵過程中,蛋白質會代謝生成胺類化合物,在適宜條件下可能生成亞硝胺[17]。因此,建立準確靈敏快速的黃酒和醬油中N-亞硝胺的檢測方法,有助于識別和預警食品安全隱患。本研究將固相萃取技術(Solid Phase Extraction,SPE)和QuEChERS法引入發酵食品黃酒和醬油N-亞硝胺檢測的前處理當中,實現復雜基質的有效凈化及N-亞硝胺化合物的高效富集,使用PTV進樣模式,采用高選擇性、高靈敏度的GC-MS/MS儀器,建立同時測定多種N-亞硝胺的方法。
1 材料與方法
1.1 儀器與試劑
TQ8040NX氣相色譜-三重四極桿質譜聯用儀(配備PTV進樣口)(日本島津公司);ME204E精密電子天平(瑞士梅特勒-托利多公司);N-EVAP-24氮吹儀(美國Organomation公司);Fotector 08HT高通量全自動固相萃取儀(睿科集團有限公司);Centrifuge 5920R冷凍離心機(德國Eppendorf公司)。
PSA QuEChERS專用超潔凈填料(40~63 μm)、GCB石墨化碳黑(120~400目)、椰殼活性炭固相萃取小柱(1 g,6 mL)。二氯甲烷、乙腈、乙酸乙酯、甲醇及丙酮(均為色譜純);無水硫酸鈉(分析純);氯化鈉(分析純)。
N-二甲基亞硝胺(NDMA)、N-甲基乙基亞硝胺(NMEA)、N-二乙基亞硝胺(NDEA)、N-二丙基亞硝胺(NDPA)、N-亞硝基嗎啉(NMOR)等N-亞硝胺混合標準品(濃度為2 000 mg·L-1,上海安譜實驗科技股份有限公司);氘代N-二甲基亞硝胺內標物(NDMA-d6,純度為98.6%,北京曼哈格生物科技有限公司);氘代N-二丙基亞硝胺內標物(NDPA-d14,濃度為1 008 mg·L-1,上海安譜實驗科技股份有限公司)。
1.2 實驗方法
1.2.1 儀器條件
(1)色譜條件。PTV進樣量:10 μL;進樣溫度:起始溫度50 ℃,保持1 min,以250 ℃·min-1升溫至250 ℃,保持8 min;普通進樣量:2 μL;色譜柱:DB-1701(30 m×250 μm,0.25 μm);柱溫:起始溫度為40 ℃,保持4 min,以5 ℃·min-1升溫至100 ℃,以20 ℃·min-1至250 ℃,保持2 min;載氣:氦氣,流速為1.5 mL·min-1。
(2)質譜條件。離子源:EI;采集模式:多反應監測(Multiple Reaction Monitoring,MRM);離子源溫度:230 ℃;四極桿溫度:150 ℃;接口溫度:280 ℃。
1.2.2 樣品處理
(1)QuEChERS法。取樣品50 g,加入內標標準溶液,混勻,加入10 g氯化鈉,加入50 mL有機萃取溶劑震蕩萃取,靜置分層,如出現乳化現象可將乳化層于6 000 r·min-1離心2 min,分出有機相并加入QuEChERS凈化試劑,渦旋3 min,離心取上清液,于35 ℃以下緩慢氮吹濃縮至5 mL,經少量無水硫酸鈉干燥后進樣。
(2)固相萃取法。椰殼活性炭固相萃取小柱依次用5 mL二氯甲烷、5 mL甲醇、5 mL水活化,取
50 g已加內標樣品上樣,重力作用自然流下,用5 mL
水淋洗,抽干10 min,10 mL有機洗脫溶劑洗脫,收集洗脫液,于35 ℃以下緩慢氮吹濃縮至5 mL,經少量無水硫酸鈉干燥后進樣。
1.2.3 標準溶液的配制
將1 mg·L-1 N-亞硝胺混合標準溶液用乙腈稀釋定容至10 mL,得到濃度為200 mg·L-1的混合標準儲備溶液。取適量NDMA-d6、NDPA-d14內標物用乙腈溶解配制成濃度為5 mg·L-1的內標儲備液。
取400 μL濃度為5 mg·L-1內標儲備液,用二氯甲烷定容至10 mL,得到濃度為200 μg·L-1內標稀釋液。取50 μL濃度為200 mg·L-1的混合標準儲備液與200 μL濃度為5 mg·L-1的內標儲備液,用二氯甲烷定容至5 mL,得到N-亞硝胺濃度為2 000 μg·L-1混合標準中間溶液(內標物氘代N-亞硝胺濃度為
200 μg·L-1)。將此混合標準中間溶液用200 μg·L-1內標稀釋液逐級稀釋并定容至1 mL,得到濃度分別為25、50、100、200、500、700 μg·L-1及1 000 μg·L-1的系列標準工作溶液。
1.2.4 加標樣品的制備
取250 μL濃度為200 mg·L-1的N-亞硝胺混合標準溶液,用乙腈稀釋定容至10 mL,得5 mg·L-1的加標母液。向50 g空白樣品中分別加入50、100、200 μL加標母液,即得濃度分別為5、10、20 μg·kg-1的加標樣品。
2 結果與分析
2.1 儀器條件的優化
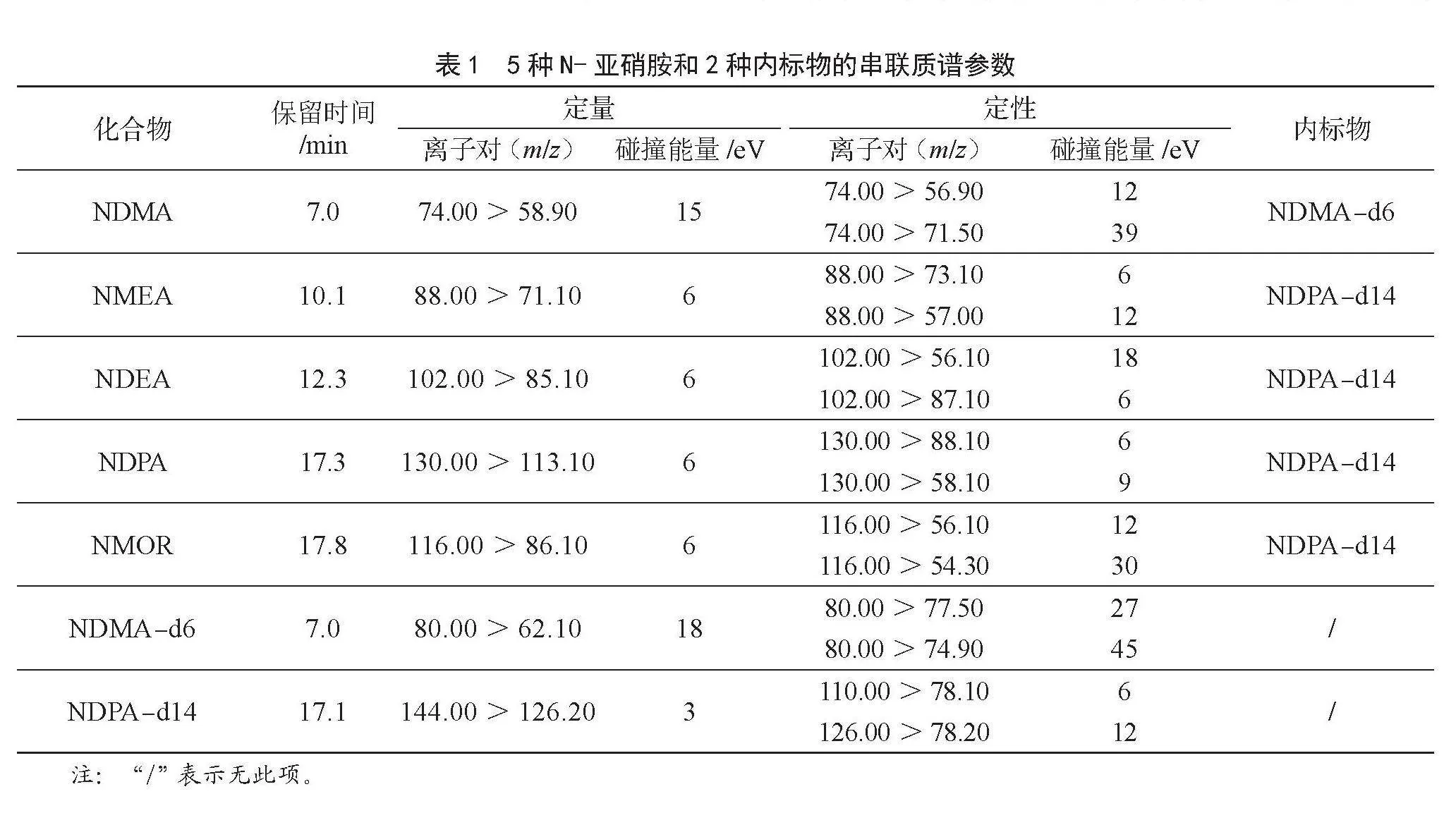
通過調整氣相色譜的參數,使用DB-1701色譜柱有效地分離了5種N-亞硝胺類化合物,實現了無雜峰干擾。按1.3.1項確定的色譜條件,對N-亞硝胺類化合物標準工作液進行全譜掃描分析,以確定用于定性和定量分析的離子對。在多反應監測模式下,對碰撞能量進行了優化,以增強定性和定量離子信號強度,優化后各化合物的質譜參數如表1
所示。
2.2 前處理方式的優化
2.2.1 QuEChERS法
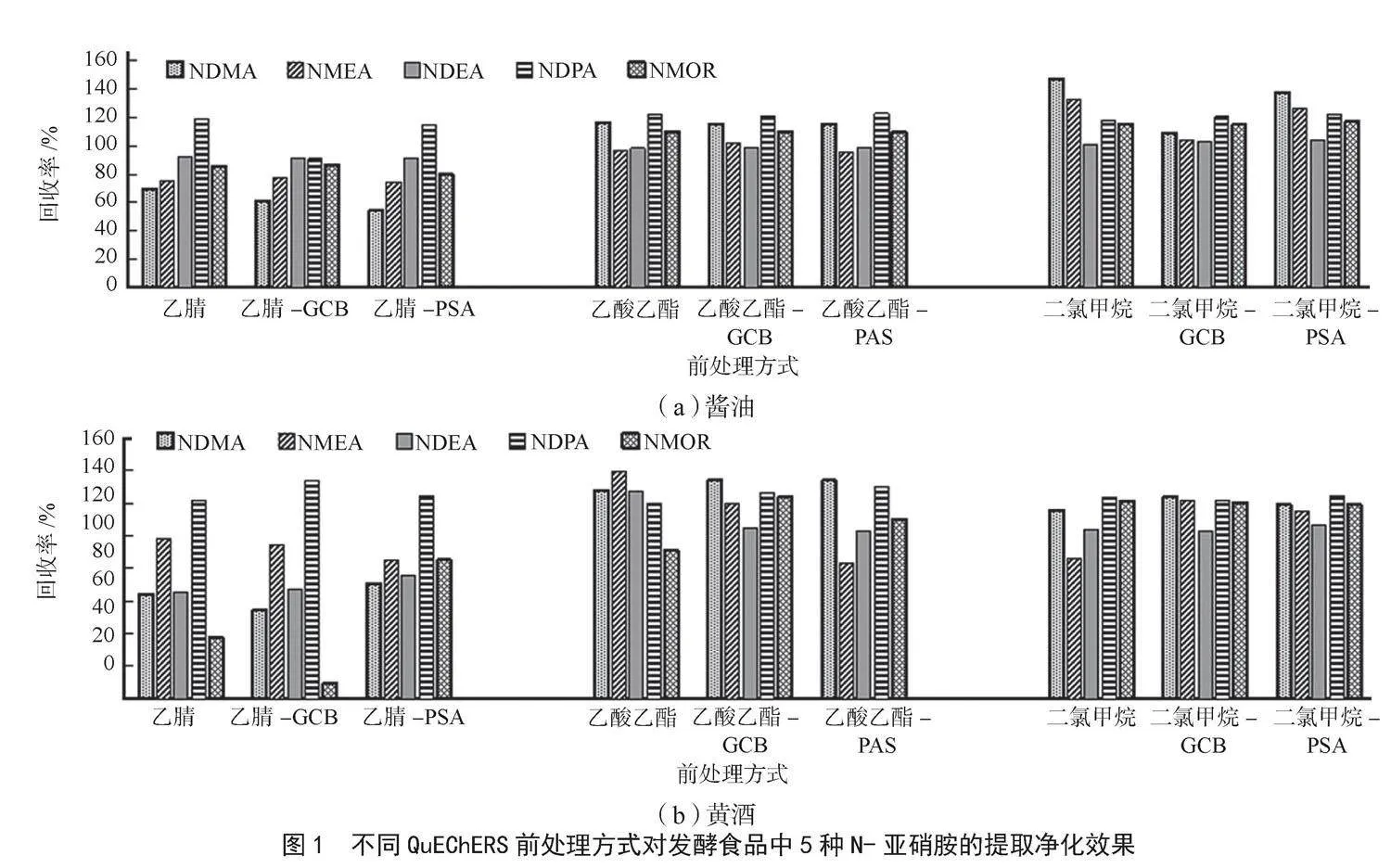
N-亞硝胺化合物因其分子質量較低且具有較強的極性,在極性溶劑中具有較好的溶解度,常見的提取溶劑包括乙腈、二氯甲烷和乙酸乙酯[18]。NaCl可以增強水相離子強度,減小N-亞硝胺類化合物在水中的溶解度,同時減少水相與有機相的乳化現象,提高萃取效率,因此在提取過程中可以加入少量NaCl。在提取過程中,有機相中混入了少量水分,所以必須先使用無水Na2SO4進行脫水處理。發酵食品黃酒和醬油中通常含有色素、氨基酸、多肽、蛋白質、有機酸及糖類等復雜基質,容易影響測定的準確性。GCB石墨化碳黑能吸附色素與非極性化合物,PSA QuEChERS專用超潔凈填料為正相硅膠鍵合吸附劑,可以吸附糖類、有機酸及少量色素。本研究對比了乙腈、二氯甲烷和乙酸乙酯3種溶劑及GCB、PSA兩種凈化劑對醬油、黃酒中N-亞硝胺類化合物的提取凈化效果,結果見圖1。
可以看出,乙腈對N-亞硝胺類化合物的提取效率最差,不同化合物的回收率差異明顯;乙酸乙酯和二氯甲烷提取效果相當。綜合考慮醬油與黃酒的提取凈化效果,二氯甲烷-GCB提取效果較好,各N-亞硝胺類化合物回收率在80%~120%,可以滿足定量分析要求。
2.2.2 固相萃取法
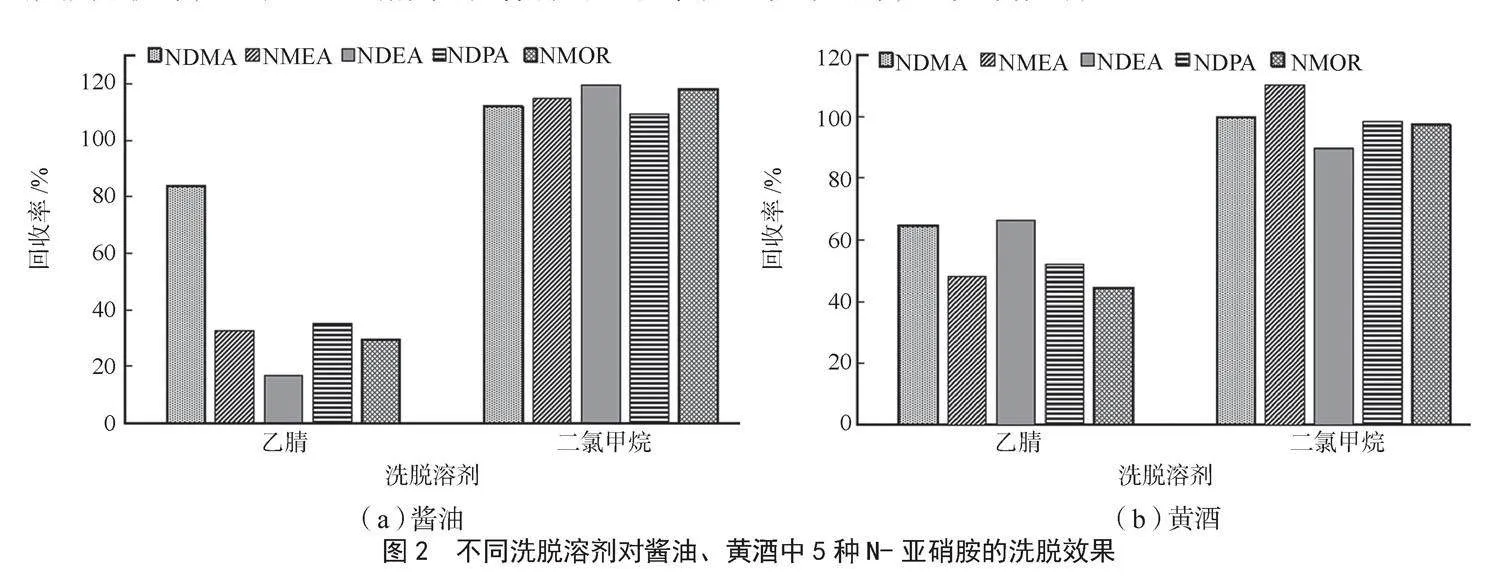
椰殼活性炭固相萃取小柱常用于N-亞硝胺類化合物的富集凈化。本研究采用加標回收實驗,對比了兩種洗脫劑(乙腈和二氯甲烷)的洗脫效果,結果見圖2。由圖可知,乙腈對N-亞硝胺類化合物的洗脫能力較差,大部分化合物的回收率低于60%,而二氯甲烷對N-亞硝胺類化合物的洗脫能力較好,5種N-亞硝胺類化合物的回收率在90%~120%。
2.2.3 前處理方式的選擇
QuEChERS法與固相萃取法對N-亞硝胺類化合物的提取富集效果相當,考慮操作步驟與操作過程中的自動化程度,選擇全自動固相萃取儀對樣品進行固相萃取前處理。
2.3 方法學評價
2.3.1 線性關系
以目標物與內標物峰面積比為縱坐標,以二者的濃度比為橫坐標繪制標準曲線,線性方程見表2。5種N-亞硝胺類化合物濃度在25~1 000 μg·L-1線性關系良好,相關系數均在0.996 0以上。
表2 5種N-亞硝胺的線性回歸方程
化合物 線性回歸方程 相關系數
NDMA y=1.227 447x 0.996 2
NMEA y=2.741 539x 0.999 3
NDEA y=1.925 023x 0.998 6
NDPA y=1.174 302x 0.998 8
NMOR y=2.082 529x 0.999 0
2.3.2 檢出限和定量限
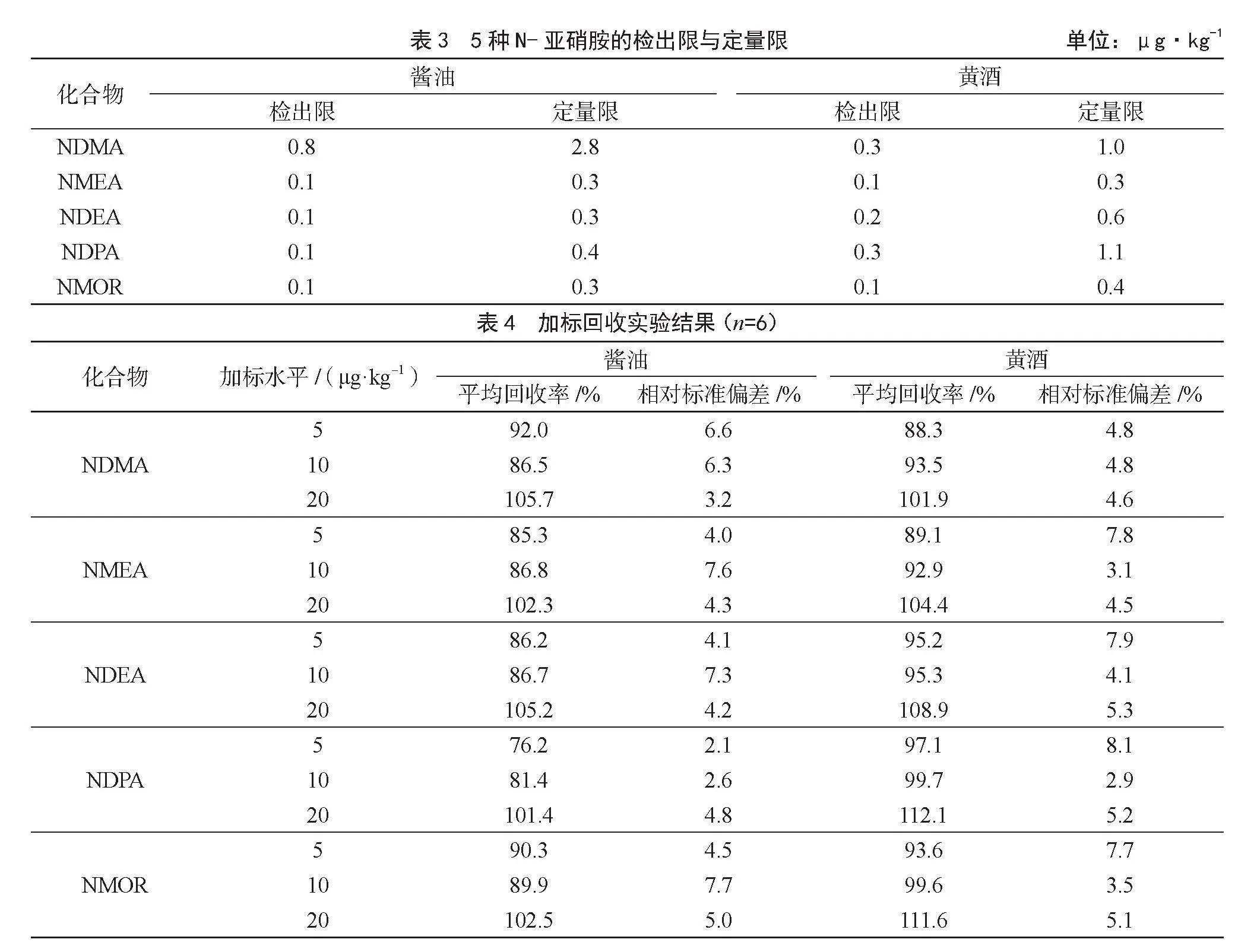
分別以超過3倍和10倍信噪比(S/N)對應的濃度確定方法的檢出限和定量限,結果見表3。在取樣量為50 g、定容體積為5 mL時,黃酒和醬油基質中N-亞硝胺類化合物的檢出限在0.1~0.8 μg·kg-1,定量限在0.3~2.8 μg·kg-1。
2.3.3 回收率和精密度
在標準曲線范圍內,選取3個梯度濃度對空白樣品進行加標,經固相萃取凈化后,進行加標回收實驗,結果見表4。由表中結果可知,5種N-亞硝胺類化合物的回收率在76.2~112.1%,相對標準偏差在2.1%~8.1%。所建立的方法能滿足醬油、黃酒中N-亞硝胺類化合物的分析要求。加標回收率與精密度結果均達到《實驗室質量控制規范 食品理化檢測》(GB/T 27404—2008)中的要求。
2.3.4 實際樣品的檢測
隨機選取市售醬油、黃酒樣品各3批次,經固相萃取凈化后,按照1.2.1項儀器條件測定樣品中NDMA、NMEA、NDEA、NDPA、NMOR等5種N-亞硝胺類化合物的含量,結果顯示僅有1批次樣品檢出NDPA,其余樣品均未檢出N-亞硝胺類化合物。
3 結論
本研究應用固相萃取技術、程序升溫進樣-氣相色譜串聯三重四極桿質譜技術,建立了一種能夠同時測定醬油、黃酒中5種N-亞硝胺類化合物的分析方法。實驗結果表明,方法的線性關系良好,檢出限與定量限較低,加標回收率及精密度均符合相關標準的規定,可以滿足日常檢測需求。
參考文獻
[1]張潔,董榕貴.QuECHERS-EMR-GC/MS測定魚肉制品中N-二甲基亞硝胺含量的方法學研究[J].貴州科學,2024,42(4):71-75.
[2]黃仁貴,馬龍,譚莎莎,等.肉制品中N-二甲基亞硝胺檢測方法的研究進展[J].糧食與油脂,2023,36
(6):1-4.
[3]伍觀保,陳文文,趙文玉,等.水中N-亞硝胺的污染狀況、來源及風險[J].環境化學,2024,43(8):2539-2554.
[4]李文慧.食品接觸用橡膠材料中的N-亞硝胺的風險分析[J].中外食品工業,2024(7):29-31.
[5]楊振,何婷婷,張忠堂,等.基于硅酸鎂前處理富集的氣相色譜-串聯質譜分析方法檢測空氣中9種揮發性N-亞硝胺[J].分析科學學報,2024,40(3):350-355.
[6]夏寒,仝凱旋,朱浙輝,等.一步式QuEChERS-氣相色譜-三重四極桿質譜法快速測定風干牦牛肉中15種N-亞硝胺[J].色譜,2024,42(5):465-473.
[7]吳翠華,杜高發,李婷,等.高效液相色譜-串聯質譜法測定腌制肉制品中的N,N-二甲基亞硝胺含量[J].食品安全質量檢測學報,2018,9(7):1695-1699.
[8]鄭鴻濤,謝國丹,譚貴良,等.低共熔溶劑-超高效液相色譜-大氣壓化學離子化-串聯質譜法測定火腿腸中9種N-亞硝胺[J].食品安全質量檢測學報,2024,15(14):52-58.
[9]王艷麗,梁秀清,陳倩倩,等.通過式固相萃取-氣相色譜-串聯質譜法測定動物源性食品中11種N-亞硝胺類化合物[J].肉類研究,2023,37(3):33-39.
[10]沈昌瑩,蘇駿敏,莫淑梅,等.QuEChERS結合GC-MS/MS測定腌臘肉制品中N-二甲基亞硝胺[J].食品工業,2023,44(7):291-294.
[11]陳婧,王立媛,胡爭艷,等.即食水產制品N-亞硝胺類化合物檢測的樣品處理方法優化研究[J].預防醫學,2023,35(8):726-731.
[12]張弛,宋瑩,潘家榮,等.氣相色譜-質譜大體積進樣法測定果汁中90種農藥殘留[J].分析化學,2015,43(8):1154-1161.
[13]蔣玲波,高卓瑤,唐雷鳴,等.QuEChERS技術結合氣相色譜-串聯質譜法同時測定水產品中10種亞硝胺[J].食品安全質量檢測學報,2022,13(1):27-33.
[14]李湖平,王靈昭,錢亮亮,等.QuEChERS結合大體積進樣法測定水產干制品中N-二甲基亞硝胺[J].食品安全質量檢測學報,2024,15(4):210-217.
[15]李登昆,張云,劉祥萍,等.固相萃取-大體積程序升溫進樣氣相色譜-三重四極桿串聯質譜測定飲用水中3種揮發性N-亞硝胺[J].色譜,2019,37(2):
216-221.
[16]姚瑞雄.分散固相萃取-同位素稀釋-氣相色譜-質譜法測定醬油中5種N-亞硝胺[J].分析實驗室,2018,37(2):183-187.
[17]謝慶超,王子,李銀輝,等.發酵肉制品中的危害因素及防控措施研究進展[J].食品科學,2023,44(19):230-238.
[18]孔祥一,莊麗麗,方恩華,等.QuEChERS-同位素稀釋-氣相色譜-串聯質譜法測定動物源性食品中9種N-亞硝胺類化合物[J].色譜,2021,39(1):96-103.
基金項目:紹興市科技局產業關鍵技術攻關計劃(2022B43003);浙江省省地協同項目(2024SDXT001-7)。
作者簡介:馬嘉俊(1992—),男,浙江紹興人,本科,工程師。研究方向:食品檢測。
通信作者:劉鎮(1986—),女,江西上饒人,博士,高級工程師。研究方向:食品檢測。E-mail: Liuz617@163.com。
猜你喜歡
中國纖檢(2016年12期)2017-01-20 09:28:19
現代農業科技(2016年20期)2016-12-20 14:51:09
現代農業科技(2016年20期)2016-12-20 09:05:36
分析化學(2016年7期)2016-12-08 00:09:44
分析化學(2016年7期)2016-12-08 00:07:08
價值工程(2016年29期)2016-11-14 01:34:54
科技視界(2016年24期)2016-10-11 18:58:00
考試周刊(2016年39期)2016-06-12 16:01:44
中國科技博覽(2016年4期)2016-04-25 07:25:47
中國科技博覽(2016年8期)2016-04-25 04:57:50
