運用孟德爾隨機化探索腸道微生物與非酒精性脂肪性肝病的因果關聯
2024-03-28 02:03:52安禎祥何遠利
安徽醫科大學學報 2024年2期
吳 閔,安禎祥,2,何遠利,3,何 松,2,劉 闖,2,孫 凱,2
非酒精性脂肪性肝病(non-alcoholic fatty liver disease,NAFLD)是臨床慢性肝病常見的病因,在沒有過量飲酒或其他慢性肝病的情況下,有5%以上肝細胞中有脂肪變性的存在,與代謝風險相關[1],并且是肝硬化和肝細胞癌的主要原因。NAFLD給患者帶來了沉重的負擔,損害了與健康相關的生活質量和工作效率。目前NAFLD的發病機制尚不清楚,但腸道微生物組與NAFLD發病有著重要關系。當下還沒有明確NAFLD的治療方法,有研究[2]表明,腸道組織可以通過腸-肝軸途徑來參與NAFLD疾病的發生發展,通過調控腸道微生物改善NAFLD患者肝臟表型。盡管腸道微生物群與NAFLD密切相關,但是其因果關系難以捉摸。孟德爾隨機化(Mendelian randomization,MR)是一種統計分析方法,主要采用遺傳變異為工具變量,觀察關聯結果推斷潛在的因果關系[3]。該研究采用孟德爾隨機化的思想,探討腸道微生物不同菌群之間與NAFLD風險的潛在因果關系。
1 材料與方法
1.1 研究設計采用兩樣本孟德爾隨機化分析腸道微生物與NAFLD之間的因果關系。以腸道微生物為暴露因素,包含211種腸道細菌性狀,結局變量為NAFLD。
1.2 數據來源使用的腸道微生物數據來自于一項多個國家的全基因組關聯研究(genome-wide association study,GWAS),該研究納入了24個隊列,18 340 例受試者,分別來自美國、加拿大、以色列、荷蘭、比利時、瑞典、韓國、德國、丹麥、芬蘭和英國,對他們進行基因測序譜和基因分型數據,通過調整年齡、性別、技術及遺傳主成分等探索腸道微生物與人類染色體遺傳變異之間的關系,課題組在5個水平(門、綱、目、科和屬)評估了211個腸道微生物群分類群。該研究基于16S rRNA基因的3個不同可變區(V4、V3-V4和V1-V2)分析了腸道微生物群的組成,并使用微生物群數量性狀基因座(microbiome quantitative trait loci,mbQTL)作圖來鑒定影響微生物分類群相對豐度的遺傳變體[4]。NAFLD基因結果關聯的數據來自FinnGen數據庫,包括894例NAFLD病例,217 898例對照。使用數據集來源詳見表1。

表1 全基因組關聯研究的細節與分析中使用的數據集
1.3 兩樣本孟德爾隨機化的假設為了保證暴露因素與結局指標之間因果關系的真實性和準確性,MR必須符合3個重要假設: ① 工具變量(single nucleotide polymorphism,SNP)與暴露因素腸道菌群強相關聯。② 工具變量SNP與結局變量NAFLD之間和其他混雜因素沒有關聯,即排除如肥胖、體重指數(body mass index,BMI)、自身免疫性疾病等混雜因素。③ 工具變量SNP僅通過影響暴露因素腸道微生物對結局變量NAFLD產生影響,不能通過其他途徑對NAFLD造成影響。F統計量一般用于評估工具變量與暴露之間的相關性強度。F統計量的計算公式為:
F應>10,如果F<10,則說明工具變量與暴露之間的關聯較弱。
1.4 工具變量選擇與腸道微生物組明顯相關的SNPs作為工具變量,參照千個全基因組項目的全基因組信息,將全基因組統計顯著性閾值參數設置為P<5×10-8。將連鎖不平衡參數設置為r2<0.001,聚集距離=10 000 kb,從腸道微生物數據中挑選出無連鎖效應的SNP,再從已選出的SNP中剔除與NAFLD有意義的SNP(P<0.05),系統收集SNPs的主要信息,包括效應等位基因、其他等位基因、β、SE和P值,用于進一步分析[5]。
1.5 孟德爾隨機化分析孟德爾隨機化方法是一種基于遺傳變異的隨機化方法,用于評估因果關系。它利用自然界中存在的基因變異,將個體分為不同的基因型,從而模擬隨機化試驗的效果,以評估某個因素對某種疾病的影響是否具有因果性。這種方法可以避免傳統流行病學研究中的混雜因素和反向因果關系,從而提高因果推斷的可信度。常使用包括逆方差加權(inverse-variance weighted,IVW)、MR-Egger、加權中位數法(weighted median, WM)、Simple Mode法、Weighted Mode法在內的高效統計方法來推斷人類腸道微生物組對NAFLD風險是否存在因果效應。使用IVW方法作為主要分析,因為它提供了最精確的效應估計,幾乎所有MR分析都將其作為主要分析。采用MR-Egger回歸檢驗潛在的水平多效性,如果截距的P<0.05,則可能存在SNP的水平多效性。MR-PRESSO檢測多效性偏差的異常值并糾正水平多效性。此外,Cochran的Q統計量用于量化所選SNP之間的異質性。為確定是否存在潛在的強影響SNP,采用留一敏感度分析法驗證因果效應估計的可靠性和穩定性。使用R軟件(版本4.0.2,TwoSampleMR、MR-PRESSO包)進行統計分析。
2 結果
2.1 孟德爾隨機化分析結果IVW分析結果表明,丙型變形菌綱(OR=0.621,95%CI=0.412~0.934,P=0.022)、普氏菌屬7(OR=0.834,95%CI=0.714~0.974,P=0.021)、脫硫弧菌目(OR=0.714,95%CI=0.519~0.982,P=0.038)3種腸道細菌性狀與NAFLD風險負相關。腸桿菌科(OR=1.481,95%CI=1.069~2.053,P=0.018)、毛螺菌科(OR=1.405,95%CI=1.036~1.904,P=0.029)、普氏菌屬9(OR=1.251,95%CI=1.025~1.527,P=0.027)、腸桿菌目(OR=1.481,95%CI=1.069~2.053,P=0.018)4種腸道細菌性狀與NAFLD風險正相關。并且篩選出來SNP的F均>10,說明工具變量與暴露之間存在較強的關聯。考慮到NAFLD易受生活方式、飲酒的影響,本實驗使用PhenoScanner查詢了陽性結果的SNP,并排除了與上述混雜因素相關的SNP。詳見表2。IVW結果森林圖見圖1。
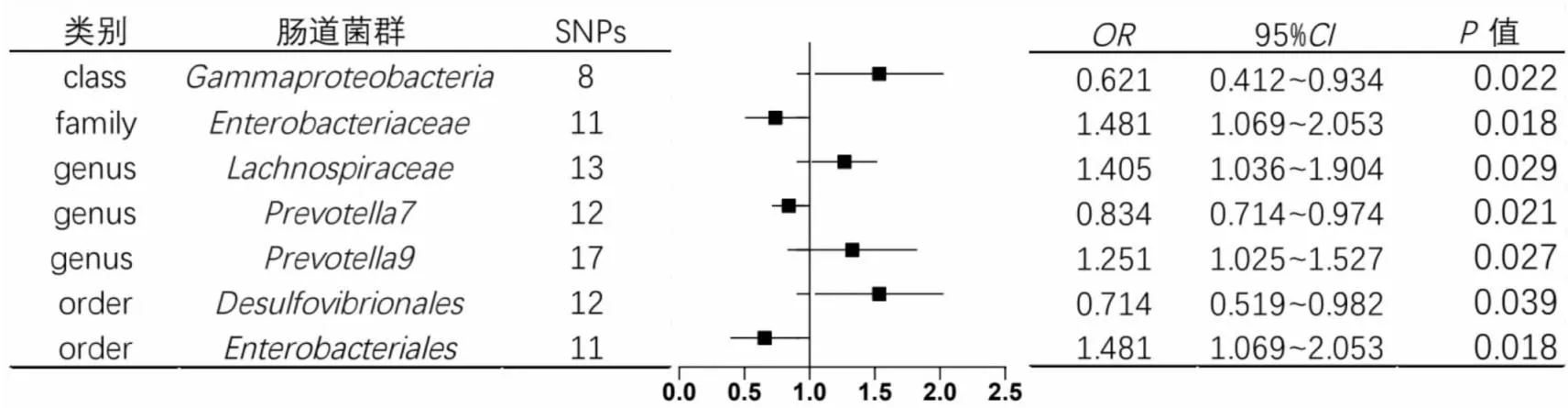
圖1 IVW結果森林圖

表2 孟德爾隨機化分析主要結果
2.2 敏感性分析Cochran Q檢驗結果顯示7種腸道細菌性狀Cochran Q檢驗P均>0.05,提示SNP間不存在異質性;多效性分析MR-Egger結果顯示7種腸道細菌性狀的P值均>0.05,因此,以上腸道細菌性狀不存在多效性水平的情況。本研究的敏感性分析采用Leave-one-out檢驗并繪制圖,留一圖表明在去除其中任何一個SNP,剩下的SNPs均在無效線的同一側,去掉任何一個SNP后對結果產生的影響均較小,則提示該MR研究結果比較穩健。散點圖則提示7種腸道菌群不同的統計方法的趨勢基本一致,說明其偏差較小。漏斗圖顯示IVW左右兩邊的點大致對稱,說明發病偏移較小。敏感性分析見表3,留一圖見圖2,散點圖見圖3,漏斗圖見圖4。

圖2 兩樣本MR結果留一圖

圖3 兩樣本MR結果散點圖

圖4 兩樣本MR結果漏斗圖

表3 敏感性分析
3 討論
本研究采用兩樣本孟德爾隨機化方法對211種腸道細菌性狀與NAFLD的GWAS數據進行因果關聯分析,共確定了7種細菌性狀與NAFLD存在因果關系,其中丙型變形菌綱、普氏菌屬7、脫硫弧菌目3種細菌性狀與NAFLD的風險成負相關,腸桿菌科、毛螺菌科、普氏菌屬9、腸桿菌目4種腸道細菌性狀是NAFLD發病的風險因素。
人類身體的胃腸道黏膜表面被數以萬計的腸道微生物所定植,這些腸道微生物組參與宿主免疫系統的調節和維持。因腸道微生物群的差異,涉及了免疫、能量、脂質和葡萄糖代謝等多種途徑,并參與了全身多種疾病的發生。近年來腸道微生物組在NAFLD中得到了廣泛的研究,有證據[6]表明,腸道微生物組-肝臟軸在NAFLD中發生作用,尤其是在纖維化和進展到晚期的疾病階段,并且該研究證明腸道微生物組中腸桿菌科的改變會影響NAFLD疾病的發展。
丙型變形菌綱是腸道微生物主要類之一,丙型變形菌綱與NAFLD等疾病有關[7],并且在NAFLD中丙型變形菌綱豐度增加。普氏菌屬是常見且豐富的專性厭氧菌,該屬的成員屬于胃腸道和呼吸道的微生物群落,并與糖尿病、心血管疾病和NAFLD等疾病高度相關,其共同特征是低度全身炎癥[8]。在非糖尿病隊列中[9],普氏菌屬豐度增加可能與胰島素抵抗有關。有研究[7]指出,患有NAFLD的兒童攜帶的普氏菌屬豐度較健康兒童高。并且使用富含普氏菌屬的生態失調微生物群從具有缺陷的炎癥小體途徑的小鼠轉移到野生型小鼠中,研究富含普氏菌屬的生態失調在NAFLD和肥胖中的作用,且該菌還加劇了蛋氨酸膽堿缺乏型飲食誘導的非酒精性脂肪性肝炎,使肝臟脂肪變性和炎癥增加以及肝功異常[10]。而另一項研究[11]則提示普氏菌屬豐度水平的降低可能對患有NAFLD的成年人有害,但毛螺菌科水平升高可能是NAFLD疾病進展的主要因素。腸道微生物中腸桿菌科是變形桿菌門的重要成員,該菌群包括許多已知在哺乳動物的大腸和小腸中定植,并且在NAFLD及結直腸癌中該細菌水平升高[12]。脫硫弧菌目菌多與神經類疾病[13]、消化系統疾病密切相關[14]。而在NAFLD疾病中脫硫弧菌目菌與瘦的NAFLD患者成負相關,與肥胖NAFLD患者呈正相關,兩者表現出相反的趨勢,但與總NAFLD呈負相關[15]。這說明腸道微生物群與疾病的發生密切相關,尤其是腸內外疾病,但其對腸內外疾病產生有益或有害作用的機制仍有待進一步研究。
雖然本研究分析了腸道微生物群,但腸道微生物群太大且異質,因此數據有限;其次,本研究雖然單獨分析了腸道微生物群與NAFLD之間的遺傳因果關系,但未進一步探索腸道微生物與NAFLD之間的生物學關系。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年18期)2018-11-14 01:48:24
汽車工程學報(2017年2期)2017-07-05 08:13:02
山東工業技術(2016年15期)2016-12-01 05:31:22
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
