高效液相色譜法測定復方異丙托溴銨霧化吸入溶液中雜質的含量
2024-03-18 14:50:28劉沁潔
理化檢驗-化學分冊 2024年2期
關鍵詞:質量
劉沁潔 ,崔 艷
(1.沈陽藥科大學 藥學院,沈陽 110016;2.上海濟煜醫藥科技有限公司,上海 200000)
世界衛生組織2019年數據顯示,全球十大死亡原因中,慢性阻塞性肺疾病(COPD)是第三大死亡原因,其死亡人數約占6%,全球疾病經濟負擔比重逐年遞增[1]。隨著全球范圍多種呼吸系統病原體的流行,特別是COVID-19在全世界范圍內的傳播[2],使得吸入制劑得到越來越多的應用和關注[3]。吸入給藥作為一種特殊的給藥方式,可以直接到達病灶部位,避免首過效應[4-6]。復方異丙托溴銨霧化吸入溶液中含有沙丁胺醇和異丙托溴銨兩種藥物成分。沙丁胺醇作為短效β2受體激動劑[7],可以擴張平滑肌,改善呼吸困難等癥狀;異丙托溴銨作為抗膽堿藥[8],有松弛支氣管平滑肌的作用。二者聯合用藥,可有效改善呼吸困難等典型COPD癥狀[9-12]。
目前只有美國藥典(USP)收錄了異丙托溴銨與硫酸沙丁胺醇復方制劑的有關物質檢測方法[13],并對3個雜質進行了質量控制。然而,研究發現:復方異丙托溴銨霧化吸入溶液中可能含有11個潛在雜質(已知雜質),主成分與雜質結構及名稱如圖1所示;USP收錄的相關檢測方法對這11個雜質的分離效果不佳,且缺少相關雜質定量方法的描述。鑒于此,本工作使用離子對試劑作為流動相[14],提出了一種不同于USP中描述的高效液相色譜法(HPLC)對11個潛在雜質進行系統分離,并對USP并未研究的制劑穩定性研究過程中降解出的8個雜質(硫酸沙丁胺醇相關雜質3, 8, 12以及異丙托溴銨相關雜質2, 6, 10, 11, 14)的定量分析方法進行驗證,用于復方異丙托溴銨霧化吸入溶液的穩定性研究以及其中這8種雜質的質量控制,同時對其中的未知雜質進行了定性及半定量分析,以期為復方異丙托溴銨霧化吸入溶液雜質檢測提供參考。
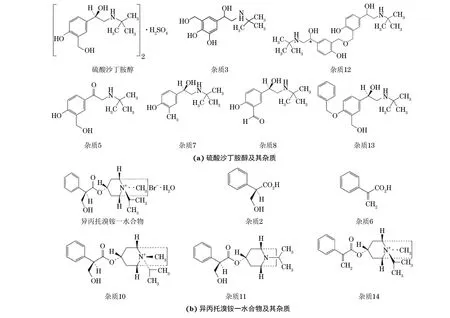
圖1 2種主成分和已知雜質的結構Fig.1 Structures of 2 major constituents and known impurities
1 試驗部分
1.1 儀器與試劑
Agilent 1260型高效液相色譜儀,配G7115A二極管陣列檢測器;XSR105DU型電子天平;Five-easy型酸度計;DK-S28型恒溫水浴鍋。
稀釋溶劑:取氯化鈉適量,加水溶解并用稀鹽酸(體積分數為10%±0.5%的鹽酸溶液)溶液調節pH至3.3,配制成0.9%(質量分數)氯化鈉溶液,備用。
系統適用性溶液:取主成分和各已知雜質對照品適量,用稀釋溶劑溶解和定容,配制成含異丙托溴銨約200 mg·L-1,雜質2;6;10;11 約1 mg·L-1,雜質14約0.4 mg·L-1,硫酸沙丁胺醇約1 200 mg·L-1,雜質3;5;7;12;13約2 mg·L-1,雜質8約3 mg·L-1的混合溶液,備用。
取適量各主成分及其雜質(USP未研究的8種雜質)的對照品,用稀釋溶劑溶解和逐級稀釋,制作的混合標準溶液系列中各化合物的質量濃度見表1。
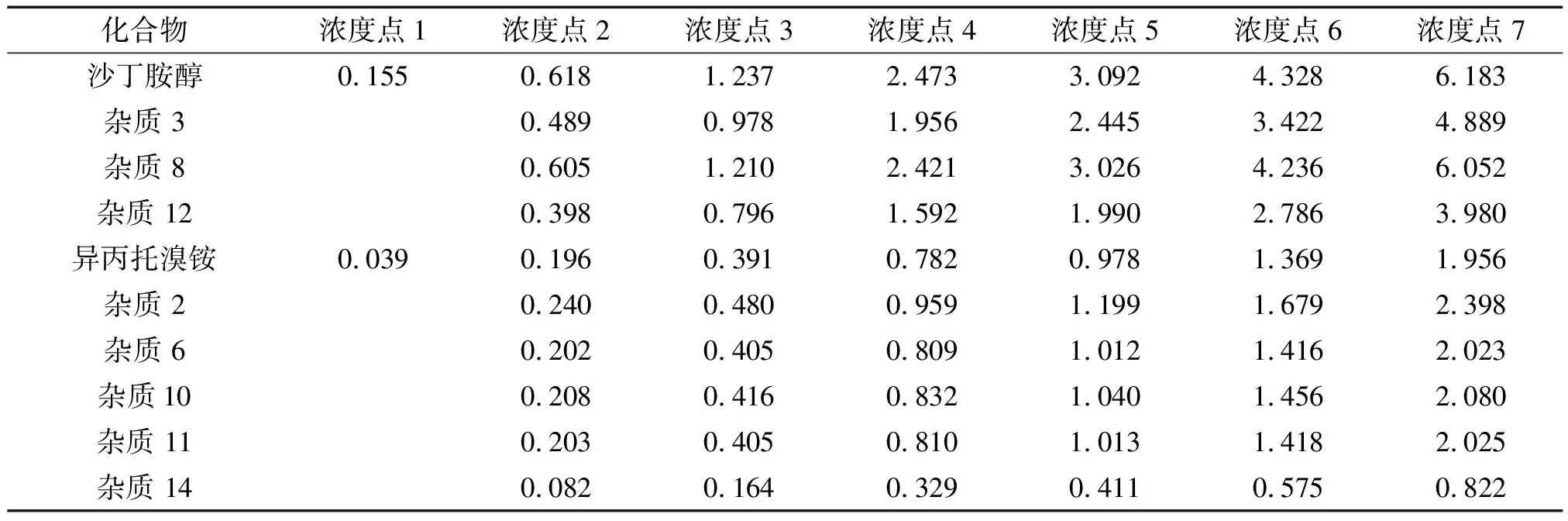
表1 混合標準溶液系列中各化合物的質量濃度
原料藥儲備溶液1:取異丙托溴銨原料藥適量,用稀釋溶劑溶解和定容,配制成含異丙托溴銨約200 mg·L-1的溶液,備用。
原料藥儲備溶液2:取硫酸沙丁胺原料藥適量,用稀釋溶劑溶解和定容,配制成含硫酸沙丁胺醇約1 200 mg·L-1的溶液,備用。
混合原料藥儲備溶液:取兩種主成分原料藥適量,用稀釋溶劑溶解和定容,配制成含異丙托溴銨約2 500 mg·L-1和硫酸沙丁胺醇約15 000 mg·L-1的溶液,備用。
混合雜質儲備溶液:取異丙托溴銨雜質和硫酸沙丁胺醇雜質對照品適量,用稀釋溶劑溶解和定容,配制成含雜質2、6、10、11約12.5 mg·L-1,雜質14約5 mg·L-1,雜質3、12約25 mg·L-1,雜質8約37.5 mg·L-1的混合溶液,備用。
雜質外標溶液:取混合雜質儲備溶液4 mL,置于50 mL容量瓶中,用稀釋溶劑定容至刻度,備用。
硫酸沙丁胺醇及其雜質3,5,7,8,12,13對照品純度均不小于99.0%,異丙托溴銨一水合物及其雜質2,6,10,11,14對照品純度均不小于98.0%。乙腈、庚烷磺酸鈉、磷酸、鹽酸、氯化鈉均為HPLC級;試驗室用水為超純水。硫酸沙丁胺醇和異丙托溴銨一水合物原料藥純度分別為99.6%和96.4%。復方異丙托溴銨霧化吸入溶液(批號20041401、20041402、20041403)均為自制,于40 ℃儲存。
1.2 儀器工作條件
Agilent?Zorbax SB-Aq C18色譜柱(150 mm×4.6 mm,5 μm);柱溫35 ℃;流量1.0 mL·min-1;進樣量70 μL。流動相A為6.4 mmol·L-1庚烷磺酸鈉溶液(用磷酸調節酸度至pH 3.8),B為乙腈。梯度洗脫程序:0~10.0 min,A為100%;10.0~60.0 min,A由100%降至75%,保持5 min;65.0~65.1 min ,A由75%跳轉至100%,保持14.9 min,檢測時間80 min;紫外檢測波長210 nm。
1.3 試驗方法
取復方異丙托溴銨霧化吸入溶液,直接按照儀器工作條件測定。
2 結果與討論
2.1 檢測波長的選擇
通過全波長(190~400 nm)掃描采集兩種主成分及其雜質對照品,以及強制降解樣品的紫外吸收圖譜,結果顯示:硫酸沙丁胺醇及其雜質在近200 nm處有最大吸收,異丙托溴胺一水合物及其雜質在近190 nm處有最大吸收;強制降解樣品中各未知雜質最大紫外吸收波長均接近200 nm;乙腈末端吸收波長約在190 nm附近[15]。綜合考慮各物質的響應與分離效果,試驗選擇的檢測波長為210 nm。
2.2 色譜柱的選擇
試驗考察了Ultimate?AQ-C18色譜柱(150 mm×4.6 mm,5 μm)和Agilent?Zorbax SB-Aq C18色譜柱(150 mm×4.6 mm,5 μm)的分離效果。結果顯示:Ultimate?AQ-C18色譜柱所得色譜圖存在異丙托溴銨拖尾,雜質分離效果不佳等問題; Agilent?Zorbax SB-Aq C18色譜柱所得兩種成分和11個雜質的分離效果均較好。因此,試驗選擇Agilent?Zorbax SB-Aq C18色譜柱(150 mm×4.6 mm, 5 μm)分離復方異丙托溴銨霧化吸入溶液中的主成分及雜質。
按照儀器工作條件測定系統適用性溶液,所得色譜圖見圖2。
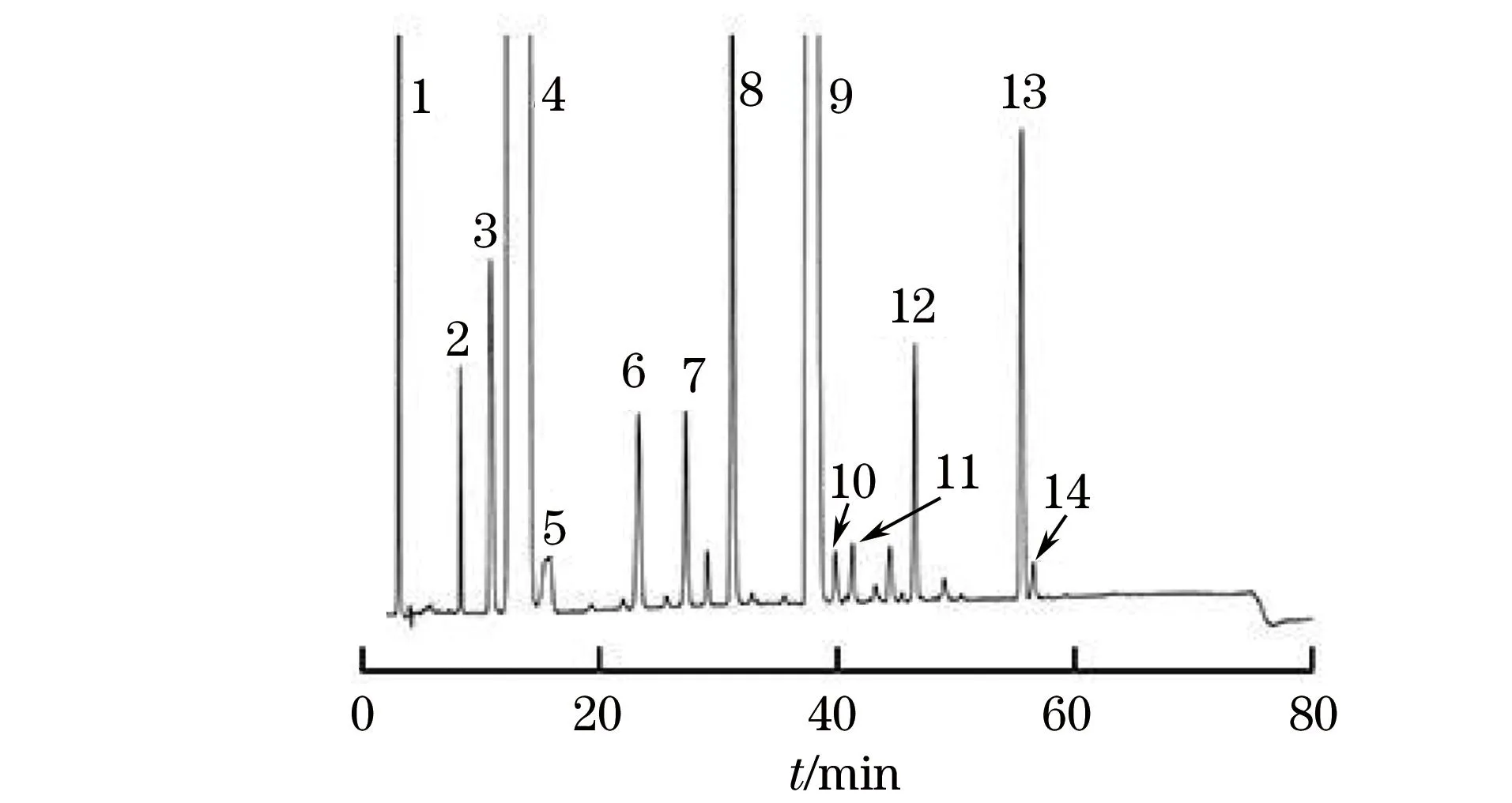
1-Br-;2-雜質2;3-雜質3;4-沙丁胺醇;5-雜質5;6-雜質6;7-雜質7;8-雜質8;9-異丙托溴銨;10-雜質10;11-雜質11;12-雜質12;13-雜質13;14-雜質14
由圖2可知,各化合物間的分離度均大于2.0,且其色譜峰峰形良好,說明各化合物可實現完全分離。
2.3 專屬性試驗
為了考察不同降解條件下方法的適用性,開展了強制降解試驗。分別取10 mL原料藥儲備溶液1,2和復方異丙托溴銨霧化吸入溶液數份,在以下5組條件下進行強制降解試驗:① 酸降解組,分別加入0.1 mol·L-1鹽酸溶液200 μL,密封,于60 ℃水浴中放置2 h,取出后各加入0.1 mol·L-1氫氧化鈉溶液200 μL中和;②堿降解組,分別加入0.1 mol·L-1氫氧化鈉溶液200 μL,密封,于40 ℃水浴中放置2 h,取出后各加入0.1 mol·L-1鹽酸溶液200 μL中和;③氧化降解組,分別加入30%(質量分數)過氧化氫溶液100 μL,密封,于室溫下放置10 h;④高溫降解組,分別將溶液密封于玻璃試管中,于80 ℃水浴中放置5 d;⑤光照降解組,分別將溶液密封于透明容器中,在冷白熒光燈和近紫外燈同時照明下放置12 d(光源總照度約為1.2×106lx·h、近紫外燈能量約為200 W·h·m-2);⑥未降解組,不做任何處理。結果見圖3。
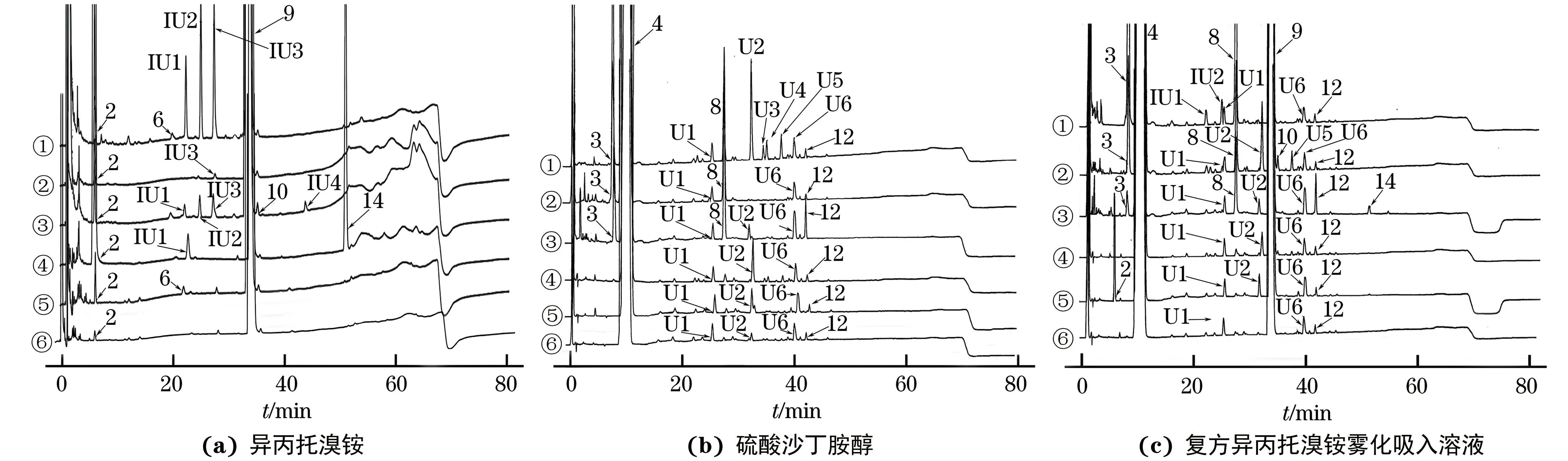
①-氧化降解組;②-酸降解組;③-光照降解組;④-堿降解組;⑤-高溫降解組;⑥-未降解組;2-雜質2;3-雜質3; 4-沙丁胺醇 ;6-雜質6;8-雜質8;9-異丙托溴銨;10-雜質10;12-雜質12;14-雜質14;IU1-雜質RRT0.67;IU2-雜質RRT0.75;IU3-雜質RRT0.83;IU4-雜質RRT1.32;U1-雜質RRT2.55;U2-雜質RRT3.19;U3-雜質RRT3.40;U4-雜質RRT3.46;U5-雜質RRT3.71;U6-雜質RRT3.96
由圖3(a)可知:異丙托溴銨單一成分強制降解試驗中的雜質主要為已知雜質2,6,10,14(雜質11降解量質量分數小于0.01%);以“RRT+相對保留時間0.67,0.75,0.83,1.32”命名未知雜質(IU1, IU2, IU3, IU4),分別為雜質RRT0.67、RRT0.75、RRT0.83、RRT1.32。推測雜質2主要由異丙托溴銨中苯基丙酰基水解產生,雜質14由異丙托溴銨上羥基消去后產生,雜質6由雜質14進一步水解或雜質2上羥基消去后產生,雜質10為異丙托溴銨氮原子手性化合物。
由圖3(b)可知:硫酸沙丁胺醇單一成分強制降解試驗中的主要雜質為已知雜質3,8,12及相對保留時間為2.55,3.19,3.40,3.46,3.71,3.96的6種未知雜質U1,U2,U3,U4,U5,U6(命名規則同上)。其中,雜質3,8由沙丁胺醇氧化產生,雜質12由兩分子沙丁胺醇脫水產生。
由圖3(c)可知:將復方異丙托溴銨霧化吸入溶液在不同降解條件產生的未知雜質按照單一成分強制降解試驗進行歸屬,主要有5種,其中主成分在氧化、光照、堿性條件下易降解。這3種條件下,異丙托溴銨中總雜質的質量分數分別為1.38%, 0.26%,1.76%;硫酸沙丁胺醇中總雜質的質量分數分別為1.16%,0.77%,0.22%;采用色譜軟件計算各化合物色譜峰純度,均不小于980[17],各化合物間分離度均不小于1.5。
在酸、堿、光照、氧化、高溫降解以及不降解條件下兩種主成分相關雜質基本一致,各化合物間均可有效分離,表明方法可以有效分離復方制劑中可能存在的潛在雜質,方法適用性良好。
2.4 標準曲線、檢出限和測定下限
按照儀器工作條件測定混合標準溶液系列,以各化合物的質量濃度為橫坐標,其對應的峰面積為縱坐標,使用不加權的最小二乘法進行線性擬合,所得各化合物的質量濃度均在一定范圍內和峰面積呈線性關系,其他線性參數見表2。
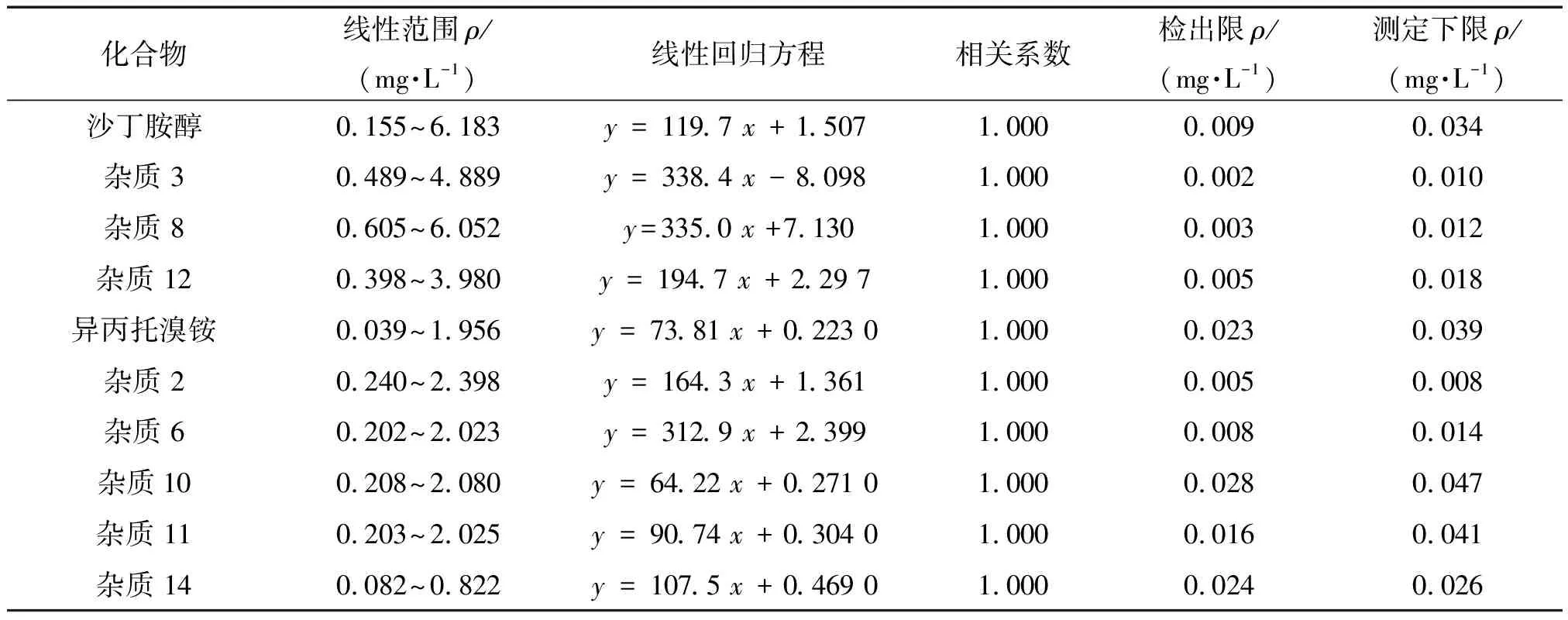
表2 線性參數、檢出限和測定下限
分別以3倍、10倍信噪比(S/N)計算檢出限(3S/N)和測定下限(10S/N),結果見表2。
2.5 復方藥物中雜質的計算
在處方濃度水平下,異丙托溴銨一水合物和硫酸沙丁胺醇在210 nm紫外波長下的吸光度差異較大,故先將兩種主成分中相關雜質分別進行歸屬后再進行計算:對于未知雜質主要依據單一主成分(異丙托溴銨或硫酸沙丁胺醇)的強制降解試驗所得的各雜質的相對保留時間(異丙托溴銨的常見4種未知雜質按照相對于異丙托溴銨的相對保留時間0.67,0.75,0.83,1.32進行定性,沙丁胺醇的常見6種未知雜質按照對于沙丁胺醇的相對保留時間2.55,3.19,3.40,3.46,3.71,3.96進行定性)進行歸屬,然后根據單一主成分面積歸一化法進行計算,以保證兩種主成分中的雜質都能得到較為準確的定量[16]。其中:異丙托溴銨響應值較小,無法歸屬的不常見雜質均按照異丙托溴銨中未知雜質計算,以達到嚴格控制雜質含量的目的;對于已知降解雜質,歸屬后按照標準曲線法分別計算其含量。
當雜質2,3,6,8,10,11,12,14通過上述方法不易獲得時,可采用加校正因子的自身對照法通過其對應的主成分計算其含量。異丙托溴銨、硫酸沙丁胺醇雜質校正因子(f)為異丙托溴銨和其雜質以及沙丁胺醇和其雜質的標準曲線斜率比,其中異丙托溴銨雜質2,6,10,11,14校正因子分別為0.4,0.2,1.1,0.8,0.7;硫酸沙丁胺醇雜質3,8,12校正因子分別為0.4,0.4,0.6。
2.6 精密度和回收試驗
取混合雜質儲備溶液2 mL,置于25 mL容量瓶中,用復方異丙托溴銨霧化吸入溶液稀釋至刻度,平行配制6份,按照儀器工作條件測定,除雜質2測定值的相對標準偏差(RSD)為1.0%外,雜質3, 6,8, 10, 11, 12, 14測定值的RSD均接近于0。
取混合原料藥儲備溶液4 mL,用稀釋溶劑定容至50 mL,作為本底溶液,按照儀器工作條件測定,用于扣除本底值;取雜質外標溶液作為外標溶液,按照儀器工作條件測定,用于計算加標量;取混合原料藥儲備溶液4 mL,分別加入混合雜質儲備溶液2, 4, 6 mL,用稀釋溶劑定容至50 mL,得到雜質濃度水平為限度值50%,100%,150%的加標樣品溶液[16],每個濃度水平平行測定3次,所得各雜質回收率,結果見表3。
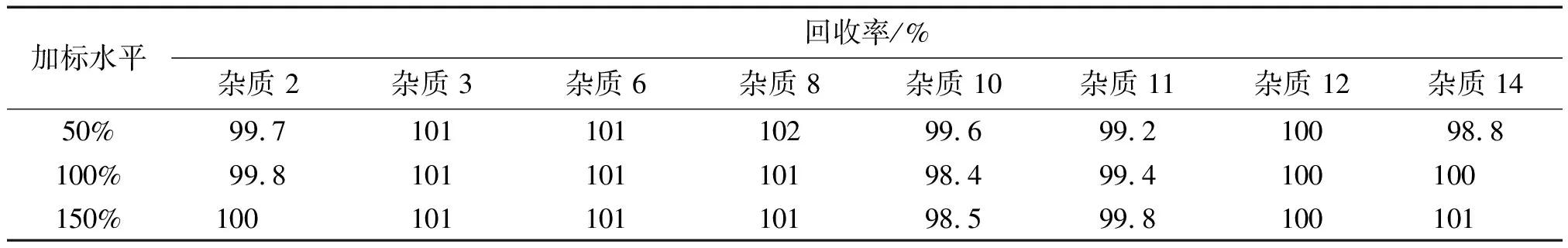
表3 回收試驗結果
2.7 穩定性試驗
取復方異丙托溴銨霧化吸入溶液約2 mL于進樣小瓶中作為樣品,在室溫條件下放置0, 11, 28 h,按照儀器工作條件測定。結果顯示,雜質2、3、6、11、14未檢出,雜質8、10、12質量分數的極差為0,0.02%,0 ,表明樣品在28小時內穩定。取雜質外標溶液于室溫條件下放置0, 28 h,放置后各化合物的相對峰面積比不小于98.5%,說明雜質外標溶液在28 h內穩定性良好。
2.8 樣品分析
按照儀器工作條件分析3批樣品,結果顯示:異丙托溴銨相關雜質10、14的檢出量均低于測定下限;雜質2質量分數分別為未檢出, 0.02%, 0.04%;雜質6質量分數均為0.03%;雜質11質量分數均為0.01%;雜質RRT1.32質量分數分別為0.05%, 0.05%, 0.03%;雜質RRT0.22(因強降解試驗中未降解出該雜質,也歸屬為異丙托溴銨雜質)質量分數分別為0.07%, 0.08%, 0.06%。硫酸沙丁胺醇相關雜質3未檢出;雜質8質量分數均為0.01%;雜質12質量分數分別為0.01%, 0.02%, 0.03%;雜質RRT2.55 質量分數分別為0.06%, 0.06%, 0.05%;雜質RRT3.19質量分數分別為0.03%, 0.03%, 0.02%;雜質RRT3.71 質量分數均為0.01%;雜質RRT3.96質量分數均為0.01%。
本工作采用HPLC測定復方異丙托溴銨霧化吸入溶液中的雜質,該方法重復性好、靈敏度高,可為復方異丙托溴銨霧化吸入溶液中雜質測定提供參考。堿、氧化和光照強制降解試驗過程中產生較大未知雜質的降解產生機理有待進一步研究。
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
