銀-二芳基乙烯化合物的合成及其光致變色性質的研究
2024-03-07 01:33:28李邵蕊徐海兵曾明華
湖北大學學報(自然科學版) 2024年2期
李邵蕊,徐海兵,曾明華,2
(1.湖北省先進有機化學材料協同創新中心(湖北大學),有機功能分子合成與應用教育部重點實驗室(湖北大學),湖北 武漢 430062; 2.廣西師范大學化學與藥物科學系, 廣西 桂林 541006)
0 引言
二芳基乙烯開關由中心的乙烯橋和兩個芳香基團組成,以優良的熱穩定性、抗疲勞性和快速光響應機制成為光致分子開關化合物中一大研究熱點。其在一定波長(λ1)的光照下,發生光化學反應,生成另一種構型的產物,再經另一波長(λ2)的照射或熱弛豫過程,又可以轉換回初始狀態[1-3]。通過引入金屬模塊來調節二芳基乙烯開關的光致變色性能是金屬分子開關的一種常見設計理念[4]。金屬離子AgI較大的半徑和多變的配位構型,使其成為理想的配位中心。通常,銀配合物中存在H鍵、Ag-Ag、Ag-C以及Ag…π等弱相互作用[5],這些相互作用一定程度上能調節配體的結構及其光譜性質。在此,本工作以咪唑為乙烯橋的二芳基乙烯開關L[6]和硝酸銀進行配位,得到[L-AgNO3]2(CHCl3)2化合物,簡稱Ag-L。通過晶體數據分析Ag-L的結構,使用紫外-可見分光光譜儀研究并對比了L和Ag-L的光致變色性質及Ag-L的抗疲勞性。
1 實驗部分
1.1 試劑
實驗所用原料和氘代試劑均在專業平臺購買。除了用于光譜測量的溶劑為光譜級溶劑,所有溶劑在使用前均經過干燥、蒸餾和脫氣處理。二芳基乙烯開關L按文獻[7]中的方法進行制備。
1.2 表征儀器
使用Bruker傅里葉-紅外分光光度計測試紅外吸收光譜。核磁氫譜通過400 MHz NMR光譜儀(Bruker Avance 400)進行測試。紫外-可見吸收光譜在Agilent Cary 6000i紫外-可見分光光度計上測定。單晶衍射數據在Rigaku XtaLAB Synergy四圓衍射儀上采集,采用Cu-Kα輻射,使用Cryalispro軟件(版本為1.171.39.34b)進行數據還原和分析。利用各向異性位移參數對所有非氫原子進行了細化。結構細化過程中使用的特殊細化如下:由于Ag-L晶體結構中存在無序,i) 使用DFIX指令限制了一些鍵長;ii) 使用ISOR指令將一些孤立原子限制為近似各向同性。配體L的CCDC號為2257692,配合物Ag-L的CCDC號為2257696,可以通過劍橋晶體學數據中心(http://www.ccdc.cam.ac.uk/data_request/cif)免費獲取。
1.3 Ag-L的制備
將二芳基乙烯開關L溶解在乙腈中,加入等摩爾的硝酸銀,回流24 h后停止反應,此時溶液為無色澄清溶液,減壓濃縮后出現少量白色固體(白色固體為未參與配位的L和過飽和析出的配合物),過濾,取濾液,旋干后得到配合物Ag-L。
2 結果與討論
2.1 單晶結構描述
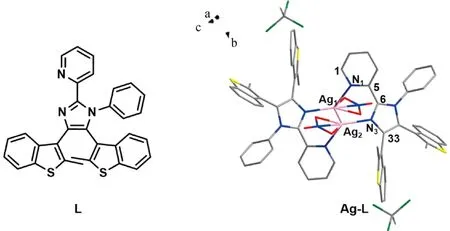
圖1 L和Ag-L的晶體結構
比較L配位前后的晶體結構,C5—C6之間的距離在與AgI配位后由原來的0.148 7 nm縮短至0.144 8 nm。配位后吡啶環中C5—N1和咪唑環中C6—N3的鍵長都發生了變化,這使得C1—N1—C5之間的角度從116.555°變為118.948°,C6—N3—C33之間的角度從106.170°減小為105.984°。L和Ag-L晶體結構的鍵長與角度變化對比如表1所示。圖2展示了不同角度的Ag-L晶體堆積圖。此外,我們還發現吡啶環上的質子與相鄰苯環之間存在C—H…π相互作用(圖2 (a))。

表1 L和Ag-L晶體結構的鍵長與角度對比
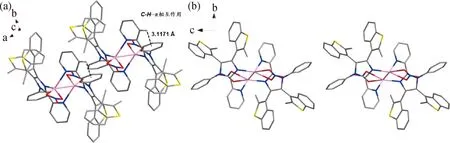
圖2 不同角度的Ag-L晶體堆積圖
2.2 紅外及核磁表征
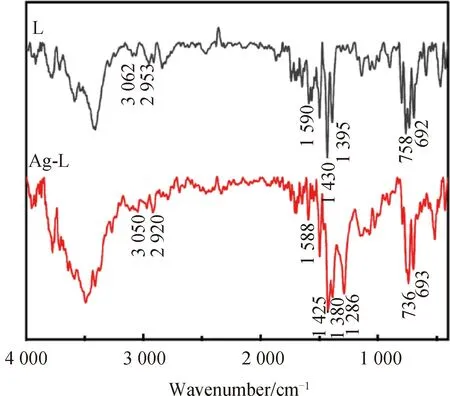
圖3 L與Ag-L的紅外光譜對比圖
將配體L和配合物Ag-L分別溶于DMSO-d6溶液并置于核磁管中,在室溫條件下進行一維核磁共振氫譜的測試。如圖4所示,配體L與AgI發生配位后,由于σ效應,L的電子云密度降低,其質子的化學位移明顯向低場移動。然而,AgI與N之間存在反饋π鍵可能會使配體的π系統電荷密度增加,導致配位后部分質子受到了屏蔽作用,向高場移動[10]。
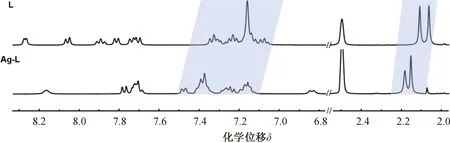
圖4 L與Ag-L的核磁氫譜對比圖(DMSO-d6, 400 MHz, 293 K)
2.3 光致變色性質研究
為了更好地研究配合物Ag-L的光致變色性質,我們將L和Ag-L分別溶解在脫水處理后的二氯甲烷中,制成濃度為1.0×10-5mol/L的溶液,通過UV-Vis光譜研究它們在累計時間下經365 nm和550 nm光照后發生的光致變色反應。
開環狀態下配體L和配合物Ag-L二氯甲烷溶液的UV-Vis譜圖中,在300 nm左右時有較強的吸收,溶液呈無色。用365 nm 的紫外光照射后,隨著光照時間的增長,L和Ag-L逐漸發生閉環反應,在約550 nm處出現一個新的吸收峰(圖5 (a)、(c)),這是閉環異構體的特征吸收,此時溶液顏色由無色變為淡紫色。可見區吸收峰強度隨光照時間增長逐漸增強,當光照到一定時間后,吸光度不再變化,此時達到光化學穩定狀態(photostationary state, PSS)。緊接著用波長為550 nm的可見光照射,閉環異構體的特征吸收逐漸減弱,直到達到PSS態,溶液由淡紫色變回無色(圖5 (b)、(d))。值得注意的是,相比于配位前的L,雖然Ag-L配合物在可見光區的最大吸收峰的波長位移變化較小,但吸收強度有非常大的提升,達到PSS態的時間也從100 s縮短至20 s。綜上,引入AgI后,二芳基乙烯化合物在二氯甲烷溶液中仍具有可逆的光致變色性質,但對其光物理和光致變色性能影響較小。
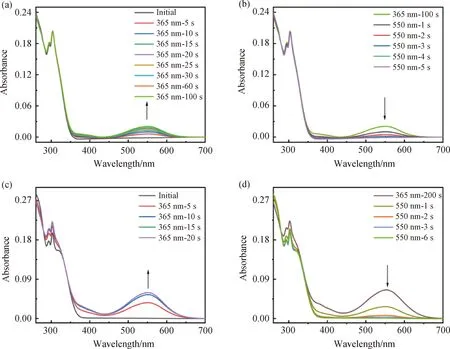
圖5 L(a, b)與Ag-L(c, d)在二氯甲烷中吸收隨光照時間變化的UV-Vis光譜
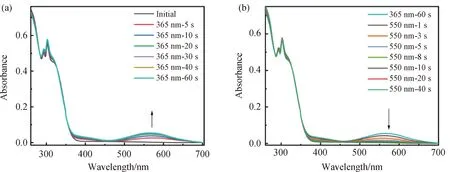
圖6 Ag-L(a, b)在PMMA膜中吸收隨光照時間變化的UV-Vis光譜
二芳基乙烯化合物的量子產率是評價其性能的一個重要參數。根據監測得到L和Ag-L開閉環過程的吸收譜圖,找出開環光穩態與閉環光穩態的最大吸收差值處波長,將該處的吸光度數值作為縱坐標,與之對應的光照時間為橫坐標,制作散點圖。再根據Y=Y0+Aexp(-kt)方程擬合曲線,得到光致異構過程的速率常數K。再根據公式(1~4),計算光致異構過程的量子產率Φ。其中,Kex是吸收在激發波長處的速率常數,σex為激發波長處的吸收截面,Ψ指的是光通量,I則是激發光源的強度[11-12]。配體L和配合物Ag-L的開閉環量子產率及其相關參數對比如表2所示。

表2 L和Ag-L的開閉環量子產率參數對比
κex=σex×Ψex
(1)
σex= (103× ln(10/NA) ×εirr
(2)
Ψex= 5 × 1015×λirr×I
(3)
Φ=κ/κex
(4)
對配合物Ag-L在聚合物薄膜中的光致變色性質進行探究。首先稱取約30 mg的PMMA于30 mL二氯甲烷溶液中,加熱使PMMA溶解,將約1 mg的Ag-L加入到完全溶解的PMMA溶液中,攪拌均勻。最后從中取適宜體積的溶液于干凈平整的玻璃板上,使溶液冷卻、溶劑揮發,制成薄膜,進行UV-Vis光譜測試。
處于PMMA薄膜中的配合物Ag-L,初始狀態呈白色,在波長為350 nm之前有很強的吸收,經365 nm的紫外光照射后,發生閉環反應,在570 nm 的位置出現新的吸收峰。該吸收峰隨光照時間的增長吸收強度不斷增大,照射60 min時達到PSS態,薄膜顏色從白色變為紫色。接著用波長為550 nm 的可見光照射,570 nm處的新吸收峰強度逐漸減弱,直到照射40 s后回到初始狀態,此時薄膜由紫色變回無色。相對于在二氯甲烷溶液中,i) PMMA中的Ag-L經紫外光照閉環后,閉環狀態的特征吸收出現輕微紅移,吸收擴展到了700 nm左右,這表明聚合物基質對配合物Ag-L的光致變色性質造成了影響。ii) Ag-L在PMMA膜中由于受到高分子介質的高粘度影響,限制了其在開閉環兩個狀態之間的自由轉換[13]。其中,開環反應在此介質中受到的影響較大,開環反應的時間從5 s延長至40 s。
2.4 抗疲勞性研究
通過UV-Vis光譜分別對配合物Ag-L在二氯甲烷溶液和PMMA膜中的光致變色過程的抗疲勞性進行研究。以初始開環狀態和365 nm輻照閉環為第一個循環,隨后通過550 nm輻照開環后再用365 nm照射為第二個循環,不斷重復光化學閉環和開環反應[14],繪制最大吸收波長處的吸光度隨循環次數變化關系的曲線。如圖7 (a)所示,配合物Ag-L的二氯甲烷溶液的光致變色性能隨循環次數的增加而緩慢衰減。相比于L[6],Ag-L在二氯甲烷中由于經受反復長時間輻照的使得金屬和配體之間的配位作用減弱,出現了少量配合物分解的狀況。而Ag-L的PMMA膜經過約10次開閉環狀態的轉化后(見圖7 (b)),閉環狀態下最大波長處的吸收沒有發生較大變化,這表明配合物Ag-L的抗疲勞性較好,具備一定的應用于光學信息存儲能力。
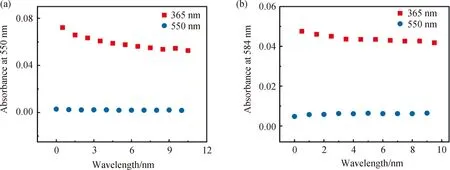
圖7 Ag-L在DCM溶液(a)和PMMA膜(b)中光致異構的抗疲勞性研究
3 結論
本工作合成了一種銀-二芳基乙烯化合物[L-AgNO3]2(CHCl3)2。運用單晶衍射儀、紅外光譜儀及核磁共振波譜儀對Ag-L結構進行了分析。在該化合物中,AgI同時與兩個不同分子上的兩個N原子結合,形成了一個扭曲的四面體。通過UV-Vis分光光譜儀對Ag-L的光致異構過程進行研究,研究表明配位后其仍然具有良好的可逆光致變色性能,并且可以通過在不同介質中調節其開關環反應速率。這為開發光子型儲存材料提供了可參考設計理念和研究思路。
