高含鹵阻燃劑污染特征及分析方法研究進展
2024-02-29 02:17:46張文睿辛正豪曾冬娟孫鵬飛孔彪梁鵬
科學技術與工程 2024年3期
張文睿, 辛正豪, 曾冬娟, 孫鵬飛, 孔彪, 梁鵬
(1.山東科技大學安全與環境工程學院, 青島 266590; 2.山東科技大學化學與生物工程學院, 青島 266590)
鹵代阻燃劑(halogenated flame retardants,HFRs)被廣泛添加于各種塑料制品、建筑材料、電子電器設備、家居產品與紡織品等中,通過增加材料耐燃性,阻止或延緩材料的燃燒[1]。由于HFRs多作為添加型阻燃劑使用,其與產品基體缺少化學鍵束縛,易在產品生產、使用及回收處置等生命全周期過程中釋放到環境中[2-3],并長時間滯留難以降解[4]。HFRs可經大氣呼吸、灰塵攝入和皮膚接觸等途徑進入人體;除此之外,進入環境中的HFRs具有持久性及生物富集性,可經食物鏈傳遞進入人體,干擾人體內分泌系統[5],并帶來潛在的神經發育毒性[6]。HFRs造成的污染及其健康效應是近20年來環境學界持續關注的前沿熱點,其中產用量最大的多溴聯苯醚(polybrominated diphenyl ethers, PBDEs)等已被列入受國際監管和限制的《關于持久性有機污染物的斯德哥爾摩公約》(POPs公約)名單,被逐步淘汰禁用[7-8],這也促使種類繁多的“理想”阻燃替代品的誕生。其中,高含鹵阻燃劑(highly halogenated flame retardants, HHFRs)因含鹵量高,阻燃性能優越,性價比突出,作為阻燃劑市場的寵兒而得到廣泛應用,最典型的HHFRs包括十溴二苯乙烷(DBDPE)、得克隆(DP)、四溴雙酚A雙(2,3-二溴丙基)醚(TBBPA-DBPE)、四溴雙酚S雙(2,3-二溴丙基)醚(TBBPS-DBPE)、三(2,4,6-三溴苯氧基)-1,3,5-三嗪(TTBP-TAZ)、三(三溴新戊基)磷酸酯(TTBNPP)等。
相比低鹵取代HFRs,HHFRs具有更大的分子體積,早期曾被認為不易在生物體內蓄積[9],因此,相關的管控法規嚴重滯后。越來越多研究證據顯示,HHFRs易在光、熱和生物酶作用下降解為低鹵轉化產物等[10],從而帶來更顯著的毒性效應[11]。然而,目前對于HHFRs環境行為及歸趨的認識相對非常有限,這與HHFRs大量的生產使用現狀不符。另一方面,由于HHFRs具有沸點高、揮發性差及高溫易脫鹵降解等特性,故以往對常規低鹵代HFRs的分析方法無法適用于HHFRs的分析測定。例如,使用氣相色譜(gas chromatography,GC)分析HHFRs需要較高的進樣口溫度及色譜柱溫度,而針對其高溫易裂解的特點則需要縮短其在高溫條件下的停留時間,即要求儀器分析時間盡可能短,這與難揮發特征不相符,給準確定性定量帶來一定的挑戰。因此,當前針對HHFRs定性定量的研究主要偏重于色譜質譜分離檢測條件、色譜柱和離子源等參數的優化,從而提升分析方法的靈敏度及檢測結果的可靠性。在此,現針對十溴聯苯醚(BDE-209)、十溴二苯乙烷(DBDPE)、得克隆(DP)、四溴雙酚A/S衍生物(TBBPA/S-DBPE)、三(2,4,6-三溴苯氧基)-1,3,5-三嗪(TTBP-TAZ)及三(三溴新戊基)磷酸酯(TTBNPP)等幾種典型HHFRs的理化性質、環境賦存、前處理方法及儀器檢測方法進行綜述,以期為開展HHFRs及其轉化產物的分析提供參考。
1 HHFRs的理化性質
表1中列舉了包括BDE-209、DBDPE、DP、TBBPA/S-DBPE、TBBP-TAZ、TTBNPP等7種典型HHFRs的基本理化性質。這些HHFRs相對分子質量介于654~1 067,其中含氯取代的DP分子量小于700,其他的分子量均在700以上。含溴取代的HHFRs有8~10個不等的溴原子,含溴量在66.2%~83.3%;而DP則有12個氯原子,含氯量為65.1%。在上述7種HHFRs中,BDE-209的沸點最低(425oC),而其他的HHFRs沸點均高于BDE-209,BDE-209高沸點難氣化的特性已有共識,可以預見這些沸點更高的HHFRs將更難氣化。此外,HHFRs辛醇水分配系數(Kow)介于8.05~12.97,表明這類化合物具有較強的疏水性,即微溶或不溶于水,從而表現出在水環境中易在沉積物蓄積的特征。由于Kow>4.5的化合物具有更高的生物累積潛力[2],這些HHFRs在水生生物體內蓄積可能帶來潛在的生態及健康風險。除此,大部分HHFRs易在紫外光或高溫下降解,如BDE-209可在紫外光條件下降解成低溴取代聯苯醚和溴代二苯并呋喃[14];DP在紫外線照射下會發生脫氯等[18],這給HHFRs特別是脫鹵產物的分析帶來一定的干擾,需更可靠的分析方法來清晰判斷脫鹵產物來源。
2 HHFRs的環境賦存
HHFRs的高疏水性使其極易吸附于有機碳或附著于大氣顆粒相中,并通過干濕沉降匯集入灰塵、土壤及沉積物中[13,17]。此外,大氣-植物交換是HHFRs從大氣環境向陸地生態系統轉移的重要途徑[16],且由于HHFRs具有親脂性,會積聚在富含脂質的物質中,并在食物鏈中被生物放大[17],因而易在動植物體內富集。普通人群主要通過室內灰塵、食物、大氣顆粒物攝入暴露于HHFRs[18]。因此,現有的研究大多關注HHFRs在土壤、沉積物、灰塵及大氣顆粒物等環境介質,以及動植物與人體內的賦存特征。表2中列舉了部分國內外相關研究中HHFR在環境介質及生物體內的賦存濃度水平。
2.1 環境樣品
通過部署在全球范圍的大氣被動采樣(global atmospheric passive sampling,GAPS)網絡,Rauert等[25]發現BDE-209在16%的大氣樣品中檢出,濃度范圍在0.8~52 pg/m3,其中BDE-209在北美地區的濃度最高,而它在歐洲地區的濃度均低于檢出限。相較于全球范圍的研究,Zhao等[26]調查了中國十個省會城市大氣中多種HFRs,發現BDE-209與DBDPE是溴化阻燃劑中占比最高的物質(>80%)。其中,BDE-209在顆粒相中的濃度介于未檢出到1.82×103pg/m3,在氣相中的濃度(未檢出到34 pg/m3)相對較低,且冬季大氣中BDE-209水平[(63±146) pg/m3]遠高于夏季[(15±27) pg/m3]。該研究報道的大氣BDE-209濃度水平與全球研究(28 pg/m3)相似[25];而大氣中DBDPE的濃度范圍在1~1 141 pg/m3,也呈顯著的季節變化特征。
作為人體暴露HHFRs的主要來源之一,灰塵樣品中也檢出了高含量的HHFRs,這些灰塵主要采集自家庭或電子垃圾拆解車間等室內場所。BDE-209作為最早一批投入使用的HHFRs,大規模的使用已導致其在環境或生物介質中普遍存在。Harrad等[24]在2008年報道了來自新西蘭、英國、加拿大、美國共計78個家庭室內灰塵樣品的PBDEs濃度,其中BDE-209的中位濃度為:英國(2.8×103ng/g)>美國(1.3×103ng/g)>加拿大(5.6×102ng/g),新西蘭作為從未直接生產或進口PBDEs的國家,在其室內灰塵樣品中雖未檢出BDE-209,但仍檢測到三至六溴聯苯醚的存在,說明含PBDEs商品的國際貿易是影響其全球分布的重要途徑。此外,在英國的兩個樣品中BDE-209檢測出了1×105ng/g和5.2×105ng/g的極高濃度,表明一些英國個人暴露于BDE 209的水平遠遠超過了通常通過飲食獲得的水平。除BDE-209外,Ballesteros-Gomez等[29]在荷蘭居民室內灰塵檢出TTBP-TAZ的濃度范圍為20~2.22×104ng/g。Guo等[31]在北美某電子垃圾回收地灰塵及大氣樣品中均檢出TTBP-TAZ,濃度范圍分別為1.17×103~4.2×104ng/g和2.23~10.3 ng/m3;同時灰塵中檢測到BDE-209、DBDPE和TBBPA-DBPE的中位濃度分別為7.9×104、1.44×103、1.58×103ng/g,TTBP-TAZ在室外環境樣品中均未檢出,說明其主要來自電子垃圾回收活動。此外,樣品中TTBP-TAZ與BDE-209等其他阻燃劑無顯著相關性,表明它們具有不同的來源或用途。相較于國外數據,Peng等[27]報道了上海居民住宅灰塵樣品中BDE-209濃度(范圍:70~1.01×104ng/g)居于全球研究中位水平,而DBDPE濃度(范圍:100~9.5×103ng/g)高于多數國家或地區。Shen等[30]在中國華南地區某電子垃圾拆解車間及當地住宅灰塵樣品中均檢出TTBP-TAZ,濃度范圍分別為nd~374 ng/g干重、4.6~431 ng/g干重。
沉積物是水生態系統中HHFRs的重要儲藏庫,對于深層沉積物的研究亦可揭示HHFRs的時間趨勢。Yang等[32]調查了美國五大湖地區沉積物中多種HFRs,其中BDE-209檢出率為83%,濃度范圍為0.87~106 ng/g干重;DBDPE檢出率為46%,濃度范圍為0.11~2.8 ng/g干重。Cai等[36]在北極西部的沉積物檢測到BDE-209、DBDPE的濃度范圍分別為nd~805 pg/g干重、nd~453 pg/g干重。Hoang等[35]檢測采集自日本西南部Beppu Bay的沉積物柱狀樣,其中BDE-209在柱狀樣0~15 cm處被檢出,濃度范圍為4.9×103~3.14×104pg/g干重,在8~9 cm處濃度達到峰值;在1991—2011年間采集的沉積層柱狀樣檢出DBDPE,濃度范圍為69~850 pg/g干重,在此期間 DBDPE 與 BDE-209 的濃度比值逐漸增加,表明這兩種化合物產用量呈相反趨勢,亦證明DBDPE作為BDE-209替代品而存在。近年來,眾多發達國家將重工業和制造業工廠遷移至發展中國家[44],由此也將污染源轉移到了亞洲、非洲等地區。Zhu等[33]發現DBDPE是珠三角表層沉積物主要污染物(范圍:1.52~1.714×103ng/g干重),并檢出DP濃度范圍為未檢出~9.46 ng/g干重,此外,該團隊還調查了中國長三角地區海底及河流沉積物樣品中常規及新興鹵代阻燃劑濃度,測得BDE-209、DBDPE、DPs的濃度范圍分別為0.038~10.7、0.176~19、0.591~7 ng/g干重[34],為珠三角及長三角地區水生態系統的HHFRs污染特征提供了依據。中國江桂斌院士及其團隊始終致力于推進環境新污染物領域的研究,該團隊Xie等[45]首次在亞洲菲律賓東北部馬里亞納海溝等深淵海溝沉積物樣品中檢測到BDE-209和DBDPE,檢出頻率為100%,且該研究發現BDE-209為16種PBDEs(范圍:179~1 220 pg/g干重)中的主要同系物,DBDPE為9種新型目標BFRs(brominated flame retardants;范圍:244~1 250 pg/g干重)中檢出的主要污染物;此外,Liu等[37]首次在中國萊州灣溴化阻燃劑工廠附近土壤檢出TBBPA-DBPE,濃度范圍在nd~1.3×107ng/g范圍,并證實醚鍵斷裂和脫溴是TBBPA衍生物形成的主要途徑。此外,工業污水的排放也是HHFRs重要的污染來源,TTBNPP化合物在日本十溴聯苯醚處理設施及其下游污水處理廠的污水樣品中被檢出[3],濃度范圍為nd~450 ng/mL,有必要進一步結合生態毒理學數據來揭示其環境風險。
2.2 生物體
由于HHFRs具有親脂性,而大氣-植物交換可使HHFRs從大氣環境向陸地生態系統轉移。鑒于松針具有較高的比表面積和脂質含量,易于捕獲HHFRs,因此Jia等[22]以松針為“被動監測器”監測上海大氣中的HHFRs。測得松針樣品中BDE-209、DBDPE和DP的濃度范圍分別為nd~324、nd~2.81、8.15×10-4~1.09 ng/g。Wang等[38]在中國南方某電子垃圾拆解地附近河岸植物樣品檢出BDE-209,濃度為3.32~76.3 ng/g干重,高于周邊農田植物樣品BDE-209濃度(14.97~18.15 ng/g),該發現表明,電子垃圾拆解活動釋放的BDE-209通過直接排放、排氣和干/濕沉積轉移到水生和陸地生態系統中,導致其在周邊植物體內富集。作為PBDEs替代品,DBDPE也在哈德遜灣和格陵蘭的北極熊及其亞種群生物組織樣本中檢出[46-47],HHFRs等新有機鹵素污染問題仍將是北極生態系統需要持續關注的環境問題。銀鷗作為五大湖水生生態系統中污染物負荷的重點關注物種已有多年歷史,Smythe等[39]調查了美國五大湖地區銀鷗生物組織樣品,測得BDE-209在脂肪樣品中濃度水平最高[范圍(623±144) ng/g脂重],其次為肌肉樣品[范圍(38.3±11.9)ng/g脂重]、腦組織樣品[范圍(16.4±4.9) ng/g脂重]和肝臟樣品[范圍(8.9±3.4)ng/g脂重]。Gauthier等[40]檢測了美國五大湖地區銀鷗蛋、反芻物及糞便樣品中的TBBPA-DBPE濃度,從上述各類樣品中得到TBBPA-DBPE濃度范圍分別為nd~497 ng/g濕重、nd~21.7 ng/g干重、nd~16.3 ng/g脂重,TBBPA-DBPE暴露與陸地食物來源相關。此外,在其早期研究中檢測到五大湖地區銀鷗蛋存在TBBPS-DBPE,但濃度低于定量限(0.3 ng/g濕重)[48]。此外,Liu等[19]檢測了渤海生物樣品中的TBBPS-DBPE的濃度,濃度范圍為nd~55.5 ng/g脂重,HHFRs在海洋食物鏈中的富集風險同樣值得關注,海產品攝入同樣是人類暴露于某些HHFRs的主要途徑。
2.3 人體賦存
盡管分子體積較大,在國內外諸多人體樣品中也發現了HHFRs的賦存。Gao等[41]在對健康人群血清中的阻燃劑濃度水平檢測中測得BDE-209、DBDPE的濃度為nd~30、nd~43.9 ng/g,研究表明健康人群在日常生活中亦暴露在高水平的HHFRs下。Liang等[42]比較了浙江溫嶺電子垃圾拆解工人與非職業居民以及城市居民頭發中的阻燃劑濃度水平,結果表明電子垃圾拆解工頭發和血清樣本中的BDE-209(46.2~724、124~2.14×103ng/g)、DBDPE(21.2~239 ng/g、26.7~440 ng/g)濃度顯著高于非職業居民(BDE-209:nd~176 ng/g、8.9~199 ng/g;DBDPE:nd~197 ng/g、4.2~127 ng/g)和城市居民(BDE-209:nd~35、nd~14.6 ng/g;DBDPE:nd~36.6 ng/g、nd~33.2 ng/g)。BDE-209是電子垃圾地區人體頭發和血清樣本含鹵阻燃劑中的主要污染物,而DBDPE 是市區樣本含鹵阻燃劑中的主要污染物。Wang等[43]調查了中國山東某BDE-209生產廠和某DBDPE生產廠工人血清和尿液樣品中HFRs。結果發現BDE-209廠工人血清中BDE-209濃度為202~5.71×104ng/g脂重,尿樣中BDE-209中位濃度為1.12 ng/mL;DBDPE廠工人血清中DBDPE濃度為87~5.44×104ng/g脂重,尿樣中DBDPE中位濃度為8.6 ng/mL,說明生產阻燃劑職業人群更易暴露于HHFRs的污染,這一結論與Matsukami等[49]和Ma等[50]的研究結果相吻合。
目前針對TTBNPP在環境及生物樣品中的檢出還缺乏相關報道,尚有待分析領域科研工作者加強相關研究。
3 HHFRs的樣品前處理
如前所述,HHFRs在污染源區域,如阻燃劑生產工廠及電子垃圾拆解回收地等地區的環境樣品中檢出水平較高,而在背景環境樣品中的濃度相對較低。因此,要對環境及生物樣品中單種或多種HHFRs進行準確定性定量分析,必要的前處理不可或缺。由于大部分HHFRs在紫外光作用下易脫鹵,因此在前處理過程中宜采用棕色的玻璃器皿進行避光處理;由于HHFRs容易吸附在各種容器及顆粒物表面造成回收率損失,故需要加入相應的內標和回收率指示物作質控。對于沉積物和土壤樣品,在樣品提取之前需進行干燥、均質和過篩,并除掉硫化物的干擾;對生物樣品中痕量HHFRs的分析,在去除樣品中所含脂質的同時,需特別注意實驗室可能存在的高污染本底值的干擾。
3.1 環境樣品
環境基質中的HHFRs的提取、濃縮及凈化與其他有機污染物類似,如圖1(a)所示,樣品提取方法包括索氏抽提、超聲萃取、加速溶劑萃取(ASE)等,ASE和超聲萃取相較于索氏提取具有萃取時間短、溶劑消耗少、提取效率高等優點。在對不同環境樣品的提取凈化中,常采用超聲萃取+固相萃取(solid-phase extraction,SPE)聯合,使用不同比例的正己烷/丙酮/二氯甲烷等混合溶劑萃取目標物,而后使用Florisil?柱、濃硫酸硅膠柱等作凈化處理[51-53]。如Ali等[51]使用超聲萃取+固相萃取的方法對50 mg灰塵中包含BDE-209、DBDPE在內的多種HFRs等進行提取凈化,先采用硅膠柱將超聲提取液中不同極性組分的HFRs進行分離,對含有BDE-209和DBDPE的組分進一步采用濃硫酸硅膠柱(44%)和弗羅里硅土(Florisil)柱凈化,該方法得到DBDPE、TBBPA-DBPE的回收率分別為94%和48%,方法檢出限均為6 ng/g。對于氣體樣品和大氣顆粒物樣品的濃縮凈化,通常采用索氏提取加SPE的方法。譬如,Zhao等[26]采用二氯甲烷對聚氨酯泡沫(polyurethane foam,PUF)和石英纖維過濾器(quartz fiber filter,QFF)所采集的氣體樣品和大氣顆粒物樣品進行24 h索氏萃取,提取液濃縮后再經多層硅膠柱凈化得到含BDE-209、DBDPE的萃取物。此外,Liu等[54]開發了基于薄層色譜(thin-layer chromatography,TLC)對高污染土壤樣品中多種TBBPA/S及其轉化產物的富集方法,該方法可經過硅膠GF254預涂TLC對土壤提取液進行分離富集,再將分離后的樣品點進行溶解后分析。與傳統的預處理技術相比,TLC分析時間更短、提取和凈化步驟更快捷。此外,Suzuki等[3]將從水和污泥樣品中得到的粗提物進行二甲基亞砜-正己烷分餾萃取,再將二甲基亞砜餾分與氯化鈉溶液混合,用乙酸乙酯萃取,得到含有TTBNPP的萃取液。
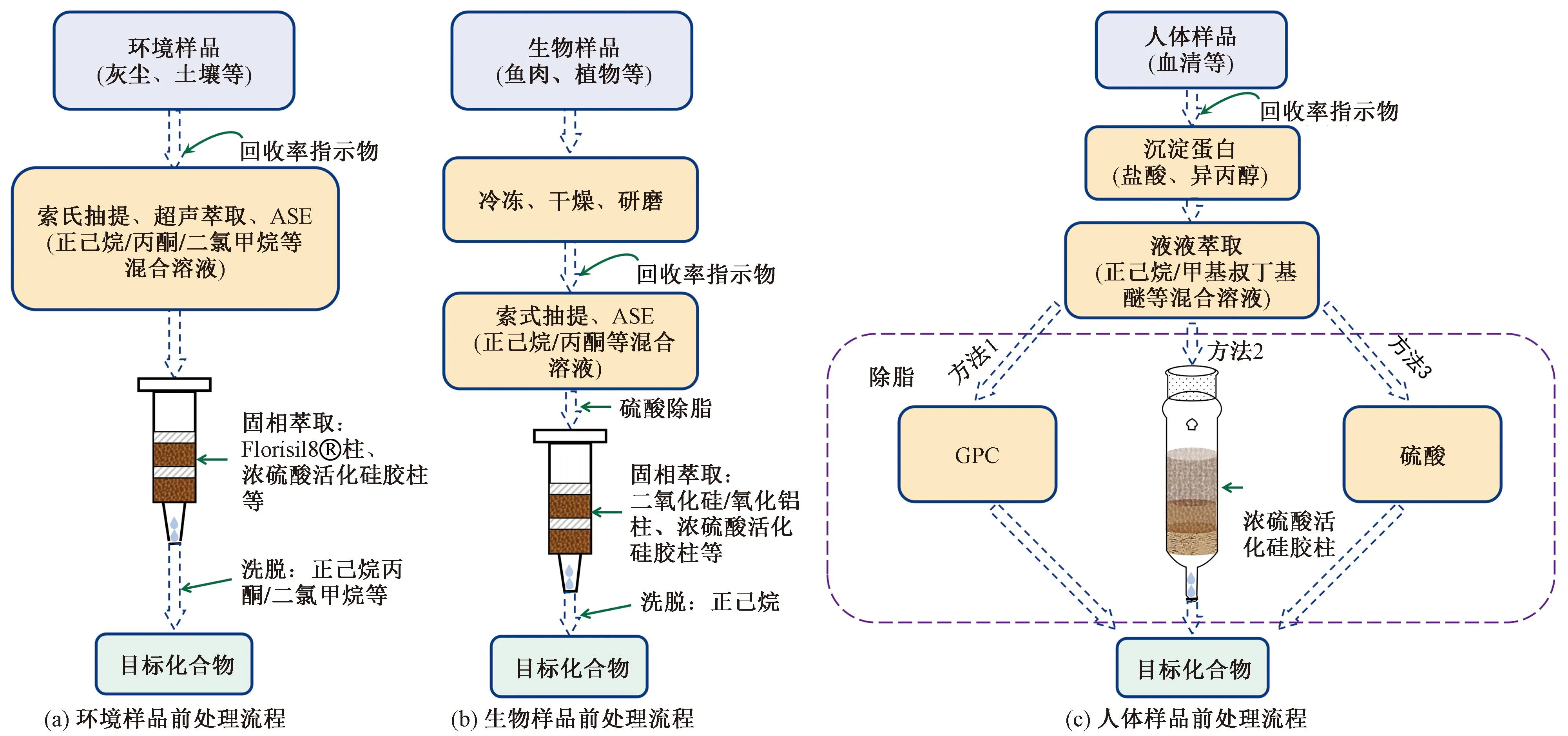
圖1 樣品前處理流程Fig.1 Sample pretreatment process
3.2 生物樣品
如圖1(b)所示,常見的生物樣品主要有生物組織和植物等。這些固體樣品通常需先冷凍干燥除水,經粉碎均質,再經索氏抽提或ASE提取。Wang等[38]將5 g植物樣品與Na2SO4混合,在索式裝置中用200 mL正己烷/丙酮(體積比1∶1)混合溶液萃取8 h,將提取物旋轉蒸發至干,并用10 mL正己烷重新溶解,旋蒸溶解過程重復3次,然后用一定量的濃硫酸除脂,收集有機相,通過多層硅膠柱濃縮至1 mL,測得阻燃劑總回收率在78.5%~117.3%,其中BDE-209檢出限為0.5 ng/g干重。Babut等[55]采用ASE通過甲苯/丙酮(體積比70∶30)混合溶液提取魚肉樣品中目標物,溶于正己烷,采用SPE的方法將正己烷提取物通過多層酸性硅膠柱,用正己烷洗脫后,得到的阻燃劑總回收率在65%~115%。此外,Cloutier等[56]針對魚肉和貽貝組織樣品開發了一種基于快速、簡單、便宜、有效、堅固耐用、安全(QuEChERS)萃取和凝膠滲透色譜(gel permeation chromatography,GPC)、SPE凈化的方法——將6 g冷凍干燥均質后的樣品與5 mL水在50 mL聚丙烯錐形管中渦旋混勻,加入10 mL乙酸乙酯后手搖1 min,然后加入鹽包(2 g NaCl和4 g Na2SO4)再手搖1 min,經離心后收集上層提取液;向管中繼續加入10 mL乙酸乙酯手搖1 min,經離心后再次收集上層提取液,將兩次提取液合并濃縮,并經由5 mL/min的二氯甲烷注入GPC,收集餾分后在正己烷中濃縮至1 mL,通過活化二氧化硅柱凈化,得到包括BDE-209在內的HFRs總回收率在87.5%~140%,方法檢出限范圍為0.03~0.65 ng/g。
3.3 人體樣品
如圖1(c)所示,對于血清等生物體液,液液萃取結合GPC、固相萃取等是最常見的前處理方式。由于GPC分離主要基于分子大小,特別適用于復雜基質條件下不同極性多種目標組分的分析,具有凈化效果好、對檢測儀器污染小的優勢,相較因極性差異而使目標物分離的固相萃取應用更廣泛。其中,Hovander等[57]開發的血漿中非極性含鹵有機物及其酚類代謝物分析的方法被各實驗室廣泛采用,通過正己烷/甲基叔丁基醚(體積比1∶1)多次萃取血漿樣品,用4 mL正己烷將提取物溶解,分別通過氫氧化鉀溶液(0.5 mol/L,50%乙醇,2 mL)和正己烷/甲基叔丁基醚(體積比9∶1,4 mL)分離出非極性化合物及酚類代謝物組分,后續的除脂可使用GPC、硅膠/硫酸活化硅膠柱以及濃硫酸等進行處理,得到多數目標物的回收率在70%~90%,該方法適合包括BDE-209、DBDPE、DP、TBBPA/S-DBPE和TTBP-TAZ等在內的大多數HHFRs的分析。此外,Covaci等[58]針對人體血清中PBDEs建立了基于OASISTMHLB(500 mg)SPE及濃硫酸活化硅膠柱凈化的方法,采用二氯甲烷洗脫,其中BDE-209回收率為98%。此外,Shi等[59]將冷凍干燥后的母乳樣品無水Na2SO4共研磨后,采用正己烷/丙酮(體積比1∶1)索氏萃取20 h,經GPC凈化和濃硫酸除脂后,得到DBDPE的回收率為70%~130%,該方法大大減少了前處理過程中所需耗材和分析污染,為人體血清中痕量分析BFRs提供了參考。
4 HHFRs的分析方法
當前HHFRs的儀器檢測方法主要是通過氣相/液相色譜-質譜法(GC/LC-MS)。GC-MS的電離方式中,相較于常用的電子電離(electron ionization,EI)[60],負化學電離(electron capture negative ionization,NCI)對具有強電負性的HHFRs有高選擇性及靈敏度,因而成為分析HHFRs的首選電離方式。NCI有時也被稱作電子捕獲負電離(electron capture negative ionization,ECNI)。在NCI模式下,目標物的電離方式是電子捕獲和反應離子化學電離。電負性分子可以捕獲帶有很低的動力學能的熱電子以產生負離子;反應離子化學電離機制則是分析物與帶負電荷的反應氣離子反應。由此,NCI屬于“軟”電離,通過更溫和的質子轉移過程產生分子離子。在使用NCI 時,由于其高選擇性,很多基質或雜質沒有響應,從而避免了干擾。目前,大部分文獻均采用GC-NCI-MS對HHFRs進行分析,由于HHFRs沸點很高,采用涂層較薄(耐高溫)的短色譜柱為更優選擇,如15 m×0.25 mm×0.10 μm規格。部分研究也報道了如何克服HHFRs在GC進樣口或色譜柱中的降解等難題,如采用程序升溫進樣口(programmed temperature vaporization,PTV)或者液體直接導入進樣口等[61]。
對于LC-MS而言,常用的電離方式有電噴霧電離(electrospray ionization,ESI)、大氣壓化學電離(atmospheric pressure chemical ionization,APCI)和大氣壓光電離(atmospheric pressure photoionization,APPI)。由于HHFRs主要是非極性化合物,使用ESI難以電離或電離效率低,通常使用APPI、APCI進行分析。在APCI模式下,樣品被噴霧到加熱室中,在電暈針幫助下使化合物在氣相中離子化;而在APPI模式下,目標分析物在氣相中吸收由真空-紫外發出的電子(10 eV或10.6 eV)后放出電子而被離子化。APCI和APPI比較適合非極性或弱極性化合物的分析,不適合熱不穩定化合物。表3中列舉了各種HHFRs典型的儀器分析方法。
4.1 BDE-209
由于CI源在檢測含有強吸電子基團的化合物時,其對負離子的靈敏度遠高于其正離子的靈敏度,使得CI源對有機鹵素具有高靈敏性和選擇性,因此GC/LC與配備CI源的各類質譜聯用是環境和生物樣品中BDE-209常用的檢測方法。Bj?rklund等[84]研究了BDE-209在GC-NCI-MS的質譜碎裂特征,發現BDE-209主要形成醚鍵斷裂的離子簇[C6Br5O]-,m/z(質荷比)范圍為482.6~492.6;其次才是Br-的特征峰m/z=79、81。因此,可利用基峰離子m/z=486.6對BDE-209進行定性定量,相比非特異性的Br-的特征離子減小了背景噪聲干擾。更重要的是可使用13C12-BDE-209做內標并采用同位素稀釋技術進行定量。當13C12-BDE-209作為同位素內標來定量BDE-209時,由于BDE-209與13C-BDE-209有重疊的m/z=488.6、490.6、492.6碎片離子,所以可以分別選擇m/z=484.6、486.6作為[12C6Br5O]-的監測離子,494.6和496.6作為[13C6Br5O]-的監測離子。基于該研究,Ma等[62]通過GC-NCI-MS分析了職業暴露人群血清樣品中的BDE-209,采用DB-5HT(15 m×0.25 mm×0.1 μm, J&W Scientific, USA)為色譜柱,以m/z=486.7、488.7作為監測離子對BDE-209進行定性定量分析,并采用13C12-BDE-209作為內標進行校正,監測離子為494.7和496.7。并且采用液體直接導入進樣口的注入方式及短色譜柱以避免GC系統對BDE-209的降解與吸附,得到的BDE-209定量限為3.6 pg/g脂重。Bu等[63]通過配備DB-5 ms柱(15 m×0.25 mm×0.10 μm, J&W Scientifific, Folsom, CA, USA)的GC-NCI-MS對不同環境室內灰塵樣品中的BDE-209進行分析,得到BDE-209的定量限為6.5 ng/g。Wang等[64]開發了氣相色譜-大氣壓化學電離源-串聯質譜法(GC-APCI-MS/MS)在MRM模式下分析人體血清樣品中的BDE-209。以13C12-BDE-209作為內標,采用DB-5MS柱(15 m×0.25 mm×0.10 μm, J&W Scientifific, Folsom, CA, USA),進樣溫度為280 ℃,1 μL進樣,BDE-209保留時間為10.49 min。由于采用APCI源,能得到大量的準分子離子,因而可以獲得比EI更高的信號強度,有助于提高檢測的準確度。與GC-NCI-MS和GC-EI-MS/MS相比,新開發的GC-APCI-MS/MS最終得到BDE-209檢出限為1.08 pg/mL,遠低于GC-NCI-MS法的50 pg/mL和GC-EI-MS/MS法(檢出限大于1 ng/mL)。基于氣質色譜手段檢測BDE-209時,GC進樣口高溫會導致一定程度的BDE-209降解,而以液質色譜檢測可以避免此類問題。Zhou等[65]采用液相色譜-大氣壓化學電離-三重四級桿串聯質譜(LC-APCI-MS/MS)在多反應監測躍遷(MRM)模式下分析廢水樣品中的BDE-209,以13C12-BDE-209為內標,采用C18柱(100 mm×2.1 mm×2.2 μm, Restek, Bellefonte, PA, USA)進行分析,得到BDE-209的檢出限和定量限分別為0.54 pg/mL和1.8 pg/mL。和常規ESI相比,APCI在靈敏度和準確度上具有較顯著的優勢,適用于多數實驗室中HHFRs的分析檢測。除了水樣,目前通過LC-APCI-MS/MS分析沉積物[78]、食品[85]等樣品中的BDE-209應用也較為廣泛。
4.2 DP
對DP及其結構類似化合物,如順式DP(syn-DP)、反式DP(anti-DP)、得克隆602(Dec-602)、得克隆603(Dec-603)以及 DP脫氯產物Cl11-DP、Cl10-DP等,通常采用GC-NCI-MS進行分析測定。Baron等[70]在選擇離子監測(SIM)式下使用GC-NCI-MS/MS對沉積物中的DP進行分析測定,以13C-syn-DP作為內標,DB-5MS為色譜柱(15 m×0.25 mm×0.1 μm, J&W Scientifific, Folsom, CA, USA),DP儀器檢出限在17~75 fg。在分析中,可選用不同色譜柱以及內標物對多種樣品中的DP進行監測,也都呈現出較高的靈敏度和分辨率。例如通過配備RTX-1614柱(30 m×0.25 mm ID×0.1 mm, Restek Inc, USA)的GC-NCI-MS對人體血清中的DP進行分析[67],以13C-syn-DP和13C-anti-DP為內標(監測離子為m/z=661.7、663.7),設置syn-DP和anti-DP的監測離子為m/z=651.7、653.7,得到DP的檢出限為0.6~1.5 pg/g脂重。分析檢測鯊魚肝臟樣品中DP及其類似物[69]時,以3′-氟-2,2′,4,4′,5,6′-六溴二苯醚(F-BDE-154)作內標,HP-5MS為色譜柱(30 m×0.25 mm×0.25 μm, J&W Scientific, Agilent),得到DP的定量限為0.4~0.8 ng/g脂重。此研究中設置離子源溫度為150 ℃,用以避免離子的高度碎裂。雖然基于GC-NCI-MS的方法有較高的特異性及靈敏度,但較低的離子源溫度使離子源受污染而頻繁清洗,導致儀器工作效率下降。由而具有高分辨率的高分辨質譜(HRMS)可以精確測定化合物的質荷比,上述研究通過GC-APCI-HRMS,還鑒定出與Dec-603結構相關的兩種新型脫氯烷類化合物,為DP類化合物的鑒定提供了新的途徑。Ayala-Cabrera等[68]采用氣相色譜-大氣壓光電離-高分辨質譜(GC-APPI-HRMS)來測定海鷗蛋樣品中DP及其相關化合物,以13C12-CB-209作為內標,采用TG-5MS色譜柱(15 m×0.25 mm×0.25 μm, Thermo Fisher Scientific, San Jose, CA, USA),設置進樣溫度為280 ℃,離子源溫度為250 ℃,乙醚作為溶劑有機摻雜劑,得到DPs的檢出限和定量限分別為1.2~43 pg/g脂重、6~140 pg/g脂重,避免了低溫下離子源的反復清潔。
4.3 DBDPE
由于DBDPE與BDE-209結構相似,主要差別僅在于苯環間的碳-碳鍵與醚鍵,因此針對DBDPE的儀器分析方法與BDE-209類似。Zhang等[72]開發了一種通過GC-NCI-MS檢測大氣可吸入顆粒物中DBDPE的分析方法,采用DB-5MS(15 m×0.25 mm×0.1 μm, J&W Scientifific, USA)為色譜柱,設置程序升溫程序以及進樣口、傳輸線、離子源溫度(分別為280、300、250 ℃),以氦氣為載氣(流速1.2 mL/min),得到DBDPE的檢出限為0.002~0.3 pg/m3。此外,Xiong等[73]基于此方法,對土壤樣品中的DBDPE進行檢測,得到DBDPE的檢出限為0.004 2~178 pg/g干重,證明該儀器分析方法針對不同環境樣品中的DBDPE分析具有良好的適用性。Klimm等[16]通過類比DBDPE和十溴聯苯(PBB-209)在紫外光作用下的脫溴特征,在GC-NCI-MS的基礎上,以DB-5HT(15 m×0.25 mm×0.1 μm, Agilent, Waldbronn, Germany)為色譜柱,通過監測Br-1的特征峰m/z=79、81以及兩個高分子量的碎片離子[M-Br]-(m/z=884.3監測DBDPE)、[M-Br+H]-(m/z=805.1監測九溴BDPEs,m/z=726.9監測八溴BDPEs),結合GC-EI-MS的質譜碎片特征,推斷并識別了3個八溴BDPEs(BDPE-197,BDPE-201和BDPE-202)和3個九溴BDPEs(BDPE-206,BDPE-207和BDPE-208),此外,通過GC-NCI-Orbitrap HRMS驗證了DBDPE含氧轉化產物(OxyTPs)的存在。和BDE-209一樣,DBDPE也會由于GC進樣口高溫發生降解,因此如何解決此問題是研究關注的要點。Neugebauer等[76]在采用氣相色譜-大氣壓電離-三重四級桿串聯質譜(GC-API-MS/MS)對云杉針中的DBDPE進行定量分析時,以13C-DBDPE作內標,DB-5HT(15 m×0.25 mm×0.25 μm, Agilent, USA)為色譜柱,分別設置進樣襯管和傳輸線溫度為280 ℃和340 ℃,盡可能提高儀器分析速度,減少DBDPE在系統中的停留時間,避免DBDPE降解。同時,在分析過程中設置梯度流量,顯著提高儀器檢出性能,得到DBDPE的儀器檢測限和方法定量限分別為0.5 pg/μL和180 pg/g干重。此外,Garcia-Bermejo等[77]建立了針對市售食品中DBDPE含量的氣相色譜-三重四級桿串聯質譜(GC-QqQ-MS/MS)檢測方法,在MRM模式下以m/z=485為前體離子,m/z=325、404分別為定量離子和定性離子監測DBDPE,采用VF-5HT色譜柱(15 m×0.25 mm×0.25 μm, Lake Forest, CA, USA),調控并優化MS操作參數、電離能量和燈絲發射電流,使分子離子特別是溴化度高的離子碎片減少,最大限度地提高目標化合物的響應水平,最終在40 eV和50 μA的組合條件下得到的DBDPE的儀器檢出限為10 pg。由于DBDPE在GC高溫程序下的熱不穩定性,研究者一直致力于開發LC-MS檢測分析DBDPE的方法,目前針對含DBDPE樣品分析檢測的LC-MS手段也具有良好的靈敏性,如Feng等[78]通過配備Waters UPLC HSS T3色譜柱(100 mm×3.0 mm×1.8 μm, Waters, Milford, USA)的LC-APCI-MS/MS對沉積物樣品中DBDPE進行檢測,在0.25 mL/min流速條件下采用線性梯度洗脫,得到的DBDPE檢出限和定量限分別為0.22 ng/g和0.74 ng/g。Zhou等[79]開發了一種HPLC-APCI-MS/MS分析水樣中DBDPE的方法,以Waters ACQUITY BEH C18(100 mm×2.1 mm×1.7 μm)為色譜柱,通過對流動相溶劑類型和比例進行優化,最終確定在流動相A、B分別為0.02 mol/L乙酸銨水溶液和甲醇/乙腈(體積比90∶10),流動相流速0.3 mL/min,柱溫40 ℃條件下獲得最優的色譜分離和靈敏度,得到DBDPE的檢出限和定量限分別為0.6 ng/L和2.0 ng/L,并且通過同位素內標13C14-DBDPE的校正提高了該方法的可重復性。由于高溴化合物在GC高溫程序下的熱不穩定性導致有時很難確保諸如DBDPE分析的準確性和靈敏度,通過溴片段進行定量也使得NCI難以區分經GC色譜柱共洗脫的含溴HHFRs,而采用配備APCI的LC-MS手段則很好地解決了這一問題,并且由于其操作簡便和高靈敏度而成為DBDPE環境分析檢測中的主流方法之一。
4.4 TBBPA/S-DBPE
GC-MS和高效液相色譜-質譜法(HPLC-MS)是常用的檢測 TBBPA/S-DBPE的方法。然而,GC-MS直接分析易導致TBBPA/S-DBPE熱分解,如Ali[51]等在開發針對TBBPA-DBPE等新型溴化阻燃劑的檢測方法中,由于GC-NCI-MS中襯管和DB-5 ms色譜柱溫度條件的影響造成目標物降解使回收率(48%)并不理想,因此GC-MS一般不是 TBBPA/S-DBPE的首選方法[86]。對于HPLC-MS,通常會引入ESI、APCI和APPI等多種離子源來滿足復雜樣品的分析需求,但它們在監測TBBPA/S-DBPE的應用中受到一定的限制,部分原因是儀器和化合物的特性造成電離困難。例如,Qu等[87]在對HPLC-ESI-MS/MS和HPLC-APCI-MS/MS的方法優化中,發現在ESI模式的全掃描優化下沒有產生任何顯著的TBBPA-DBPE前體離子,而APCI模式下雖然可獲得TBBPA-DBPE的特異性光譜,但其靈敏度不足以檢測TBBPA-DBPE在實際環境樣品中的濃度。Letcher等[80]開發了一種通過LC-APPI-MS/MS在APPI負模式下檢測TBBPA/S-BDPE的分析方法,其中LC配備ACE 3 C18分析柱(2.1 mm×50 mm×3 μm)和ACE 3 C18保護柱(2.1 mm×10 mm×3 μm, Aberdeen, UK),流動相為水和丙酮,流速為0.2 mL/min,流動相梯度如下:0%丙酮相于進樣后0.01 min升至100%丙酮相,保持15 min,再于1 min升至100%水相,保持14 min。該梯度下降低了背景噪聲,從而提高了信噪比,使得TBBPA/S-BDPE色質譜均獲得了良好的峰形。通過MRM模式實現對TBBPA/S-BDPE的高靈敏度定量,其中TBBPA-DBPE的監測離子為m/z=975.5、79,TBBPS-DBPE的監測離子為m/z=997.4、79,得到的TBBPA-BDPE方法檢出限和定量限分別為0.07 ng/g脂重和0.25 ng/g脂重,儀器檢出限為32 pg;TBBPS-DBPE方法檢出限和定量限分別為1.28 ng/g脂重和4.27 ng/g脂重,儀器檢出限為112 pg。APPI雖然對TBBPA/S-BDBPE檢測靈敏且具有特異性,但需要額外相匹配的摻雜劑用于電離[88],難以適應大多數分析實驗室的日常需求。因此針對TBBPA/S-DBPE儀器分析技術的開發也面臨巨大挑戰。
目前,基于高效液相色譜-二極管陣列檢測器(HPLC-DAD)立體化檢測TBBPA/S-DBPE得到開發和應用,尤其在定性和定量方面發揮極大優勢。Liu等[19]采用HPLC-DAD和HPLC-MS/MS串聯ESI源分別檢測螺類和魚類等海洋生物中的TBBPA-DBPE、TBBPS-DBPE,以D10標記的TBBPA(10 ng)作為內標,ZORBAX ODS為色譜柱(150 mm×3 mm×5 μm, Agilent),得到的TBBPA-DBPE檢出限和定量限分別為6 ng/g干重和20 ng/g干重,儀器檢出限為3 ng;TBBPS-DBPE檢出限和定量限分別為20 pg/g干重和60 pg/g干重,儀器檢出限為1 pg,這也是首次報道在生物樣品中檢測到TBBPS-DBPE。隨后該課題組首次建立了一種基于薄層色譜(TLC)樣品預處理和HPLC-DAD(UV=214 nm)相結合的新方法[54],可用于測定含TBBPA-DBPE和TBBPS-DBPE的土壤樣品。選用Zorbax ODS柱(150 mm×3 mm×5 μm, Agilent)為色譜柱,以乙腈和水(含0.1%甲酸)為流動相,流速為0.6 mL/min時,分離效果最好,且TLC具有更高的樣品通量、更短的分析時間和更少的提取和清理步驟,為后續HPLC-DAD的檢測有效的消除了干擾,得到TBBPA-DBPE和TBBPS-DBPE的檢出限分別為36 ng/g和49 ng/g,定量限分別為120 ng/g和160 ng/g,該法快速且經濟高效。除此之外,HPLC-DAD聯合其他技術在環境及生物樣品中檢測TBBPA/S-DBPE得到廣泛應用[89-90],也成為目前檢測TBBPA/S 及其衍生物的重要手段之一。同時,對于TBBPA-DBPE轉化產物,可采用超高分辨率傅里葉變換離子回旋共振質譜(FTICR-MS)進行初步篩選[37],利用超高性能液相色譜-軌道阱高分辨率質譜(UHPLC-Orbitrap HRMS)對污染區域土壤樣品進行定性和定量分析,以13C12標記的TBBPA作為內標,得到的目標轉化產物檢出限為0.02~3 ng/g干重。該方法具有高靈敏度和良好的可重復性,可用于其他復雜的生物和環境基質中TBBPA/S-DBPE的分析。
4.5 TTBP-TAZ
在已有研究報道中,氣質色譜和液質色譜均可對TTBP-TAZ進行分析檢測。Guo等[31]采用GC-EI-HRMS對灰塵中TTBP-TAZ進行分析時,采用RTX-1614為色譜柱(15 m×0.25 mm×0.1 μm, Restek Corporation, Bellefonte, CA),以BDE-118為內標,得到TTBP-TAZ的檢出限為0.6 ng/g。作者通過EI-HRMS掃描樣品時,根據TTBP-TAZ結構分析,該化合物可以失去一個溴原子形成以m/z=987為中心的離子簇,或失去其三個溴化環(R=C6H2Br3)中的一個形成以m/z=754 為中心的離子簇。但對于TTBP-TAZ這種低揮發性高分子量化合物,在使用GC-MS測定時,對樣品的前處理要求高,且出現結果不穩定的現象。因此,目前液質是測定TTBP-TAZ的主流方法。Baesteros-Gomez等[29]開發了一種基于直接探針與大氣壓化學電離-高分辨率飛行時間-質譜(APCI-TOF-HRMS)結合的方法,通過直接探針可將固體樣品直接引入 MS 源,省去樣品制備和色譜分離步驟,可以對環境樣品進行快速篩查;同時,采用溶劑萃取法,通過LC-APCI-TOF-HRMS確認篩選結果,并定量測定塑料和粉塵樣品中TTBP-TAZ的水平,得到TTBP-TAZ的檢出限和定量限分別為20 ng/g和60 ng/g。這個檢測方法也在后續有關WEEE污染的聚合物玩具材料中TTBP-TAZ口腔遷移研究中得到了應用[91],此外,ESI離子源對TBBP-TAZ進行測定可以得到較好的電離效果。Shen等[30]采用HPLC-ESI-MS/MS在MRM模式下檢測電子垃圾回收區的灰塵樣品,TTBP-TAZ檢出限和定量限分別3.8 ng/g干重和12.7 ng/g干重。MRM 模式選擇性和抗干擾能力強,靈敏性高,可直接將樣品過濾后上機測試,無需再用有機溶劑提取轉移。而LC-MS/MS 可通過靈活調節離子化模式來測定TTBP-TAZ,如Lorchner等[21]采用LC-ESI-MS/MS對沉積物中的TBBP-TAZ進行了分析,以13C18-TTBP-TAZ作內標,選用C18為色譜柱(100 mm×2 mm×3 μm, Phenomenex?, Aschaffenburg, Germany),總運行時間為35 min。在使用該方法時,需要對色譜方法進行優化,選擇水(0.1%甲酸)和甲醇/乙腈(80∶20)溶液為兩種流動相,設置柱溫40 ℃、流速0.25 mL/min時可得到最好的峰形和分離結果,TTBP-TAZ檢出限和定量限分別為10.2 ng/g和27.8 ng/g。這樣可避免嚴重的基質干擾,極大提高靈敏度。
4.6 TTBNPP
作為一種新型HHFRs,目前關于TTBNPP相關研究報道較少。在采用氣質聯用方法進行測定時,不同離子源模式下的檢測效果差異較大。Halloum等[92]發現,在EI正模式的全掃描MS中,未檢測到TTBNPP的分子離子,TTBNPP在m/z=145處檢測到碎片離子,推斷為[C5H6Br]+,具有特異性的峰為丟失一個側鏈的m/z=712.6;而在APCI正模式的MS中可檢測到TTBNPP的[M+H]+離子以及失去一個溴的[M—Br]+離子。在兩種離子源串聯GC-MS/MS對加標魚類樣品的檢測中,EI未得到任何TTBNPP可供分析的結果,而APCI檢測TTBNPP得到的相對標準偏差值為43%,說明該檢測方法無法獲得TTBNPP的準確定量結果。而采用液質聯用方法能夠對環境樣品中的TTBNPP進行定性和定量分析。McGoldrick等[81]采用配備C18柱(100 mm×2.1 mm×3.5 μm)的LC-ESI-MS/MS在正離子模式下對魚樣中的TTBNPP進行檢測,雖然并未在實際環境魚樣中檢測到TTBNPP,但通過加標實驗得到的TTBNPP檢出限與定量限分別為0.16 ng/g脂重和0.49 ng/g脂重,為檢測TTBNPP提供了一種良好可重復的方法。Santin等[82]采用LC與配備ESI源的四極桿線性離子阱串聯質譜(QqLIT-MS/MS)結合的方法在SRM模式下對魚類樣品中的TTBNPP進行了分析,色譜柱采用RP-18柱(125 mm×2.0 mm×5 μm, Merck, Darmstadt, Germany),流動相為水(0.1%甲酸)和甲醇(10 mM醋酸銨),最終得到TTBNPP的檢出限和定量限分別為37.4 ng/g脂重和125 ng/g脂重,儀器檢出限和儀器定量限分別為58.8 pg和196 pg。Lu等[83]開發了一種超高效液相色譜-大氣壓化學電離-串聯質譜(UPLC-APCI-MS/MS)在APCI正離子模式下檢測海鷗蛋中TTBNPP的方法,以C18柱(50 mm×2.1 mm×1.6 μm, Phenomenex, CA, USA)為色譜柱,在MRM模式下通過監測離子m/z=1 018.14、226.89、144.96對TTBNPP進行定性定量,得到TTBNPP的檢出限和定量限分別為0.02 ng/g脂重和0.1 ng/g脂重,顯著低于LC-ESI-MS/MS。該方法通過使用UPLC使得待測組分獲得更高的分離度和靈敏度,與LC相比在含TTBNPP樣品的痕量分析中更具優勢。
5 結論與展望
已有研究表明,HHFRs產用量逐年增加,在各類環境介質中普遍存在(圖2),勢必會對生態環境和人群健康帶來較大的潛在威脅。但目前國內外對其環境行為歸趨和風險的研究尚未全面展開,仍存在以下問題。
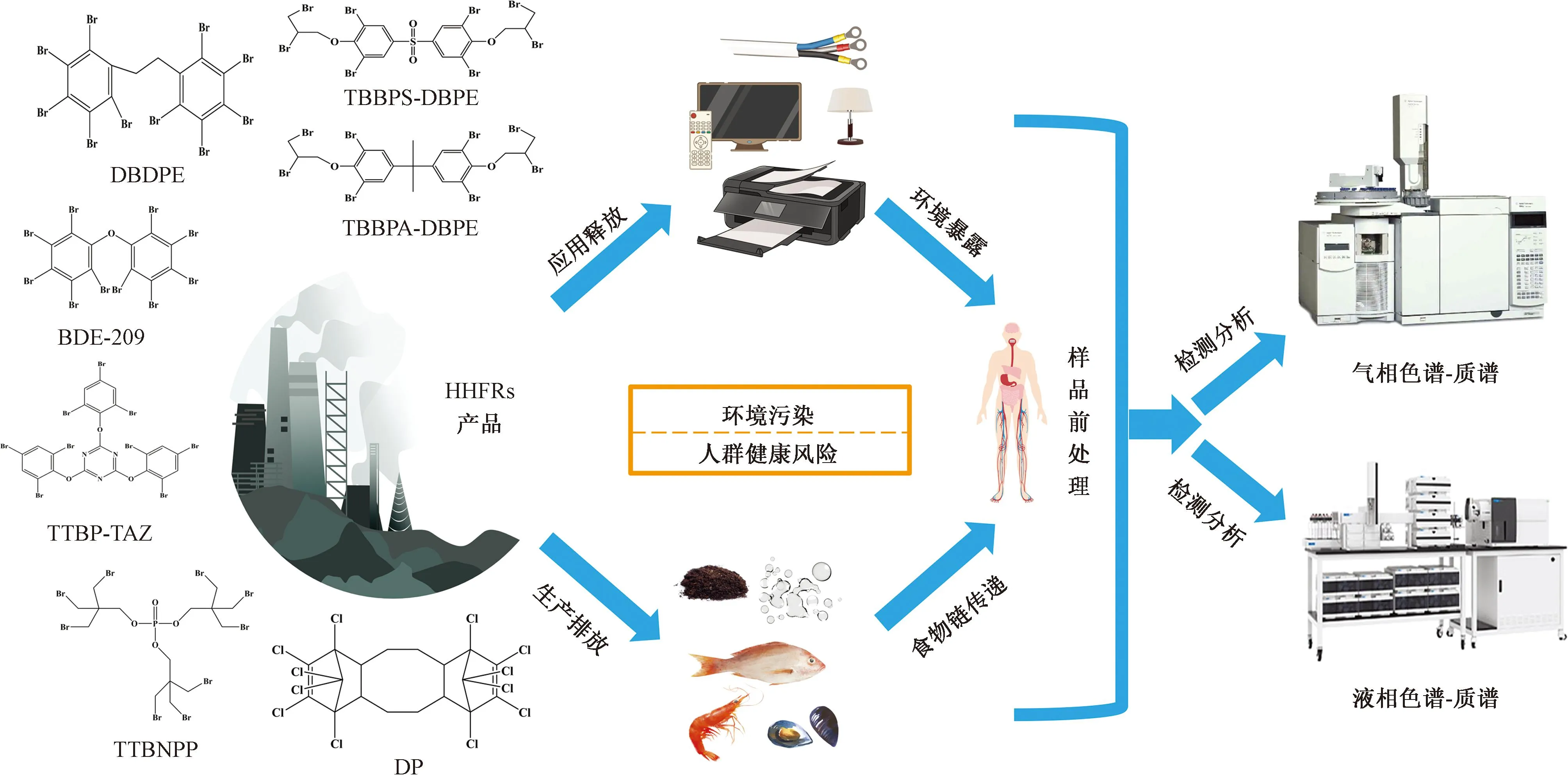
圖2 HHFRs研究概括圖Fig.2 Research summary of HHFRs
(1)已報道的HHFRs數據主要圍繞北美(美國、加拿大)、歐洲和東亞(中國、日本、韓國)的少數國家,而主要電子產品的進口國如南美、俄羅斯、印度等地以及許多發展中國家的HHFRs數據報道相當有限;BDE-209、DBDPE和DP等傳統的HHFRs受到關注較多,是環境介質及人體內檢出率及濃度較高的HHFRs,但有關TBBPA/S-DBPE、TBBP-TAZ、TTBNPP的研究有待進一步展開。
(2)對于前處理技術,傳統HFRs的SPE、液液萃取等方法也適用于HHFRs的提取與凈化,通過對萃取溶劑和吸附填料的選擇和優化能實現較好的萃取率。但HHFRs和一般HFRs在物化性質上存在較大差異,傳統前處理技術對多種HHFRs在萃取效率、樣品通量以及復雜基質條件下的萃取還存在較大缺陷。
(3)對于儀器分析,當前主流的分析手段是將HHFRs與其他HFRs通過氣質或液質聯用進行同步分析,這樣較單一分析方法更節省時間和成本。但由于HHFRs的特質以及主流儀器分析手段存在的優缺點,如何實現同步分析和表征復雜樣品中可能存在的全部HHFRs及其轉化產物對研究者來說是很大的挑戰。
(4)在氣質聯用中,NCI對大多數HHFRs的敏感性使其成為目前質譜中常用的電離首選。然而由于多數含溴HHFRs在配備NCI的MS中主要通過產生溴片段作為信號用于定量,因此NCI不能區分與GC色譜柱共洗脫的含溴HHFRs,MS檢測器中選擇性的缺乏導致想要在GC色譜柱中更好地分離HHFRs的難度增大。
(5)對液質聯用而言,APPI和APCI是分析HHFRs的主要電離技術,而APPI對紫外燈和摻雜劑的要求使其在大多數分析實驗室中的適用性相當有限。如何基于APCI的液質手段,實現在單份復雜基質樣品中,將待測HHFRs目標組分有效分離出峰將是下一步方法開發中的巨大挑戰。
考慮到HHFRs與一般HFRs性質的差異,如HHFRs的高度疏水性、低揮發性和難電離等特點,并結合目前的研究現狀和存在的問題,今后相關的研究應集中在下面幾個方面。
(1)結合ASE、QuEChERS等高效的新型前處理方法,開發提取時間短、溶劑消耗少、分離凈化步驟簡單的復雜基質中多種HHFRs前處理技術;建立質控體系,提高不同實驗室研究數據間的可比對性。
(2)對于分析方法,既要開發適用于精準定性定量分析的常規方法,通過優化色譜質譜分析檢測條件、色譜柱和離子源等參數以提升分析方法的靈敏度和結果的可靠性;又要結合先進手段如直接探針(DP)方法與APCI-TOF-HRMS相結合的快速篩選方法,對環境介質中的HHFRs污染源進行快速識別。
(3)系統開展HHFRs轉化產物的分析,亟需合成更多的HHFRs轉化產物單體以及相應的同位素內標,并且能夠通過商品化途徑獲取,以便國內外學者針對HHFRs的環境歸趨及毒性效應等方面開展更深入的研究,為準確評估其潛在生態和健康風險提供科學依據。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
